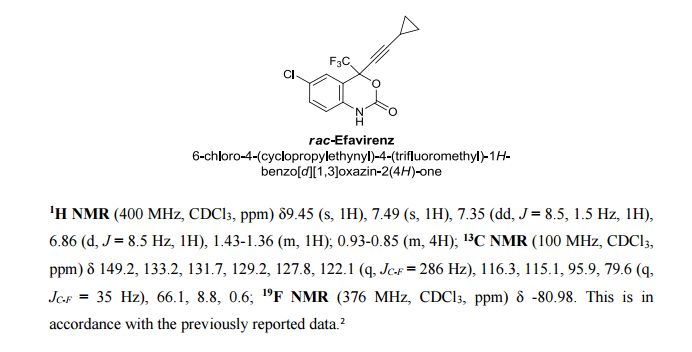
![STR1]()
![Tenatoprazole.svg]()
Tenatoprazole
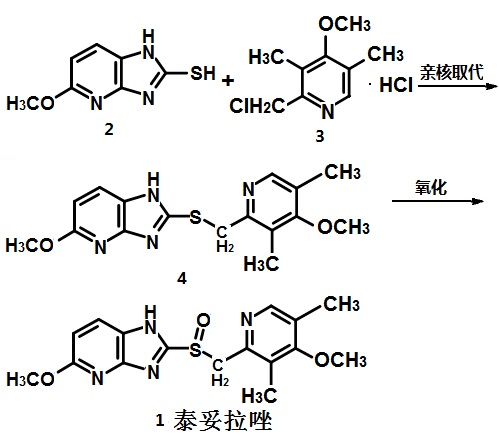
泰妥拉唑
| Tenatoprazole; 113712-98-4; Ulsacare; Protop; TU 199; TU-199; |
| Molecular Formula: |
C16H18N4O3S |
| Molecular Weight: |
346.40412 g/mol |
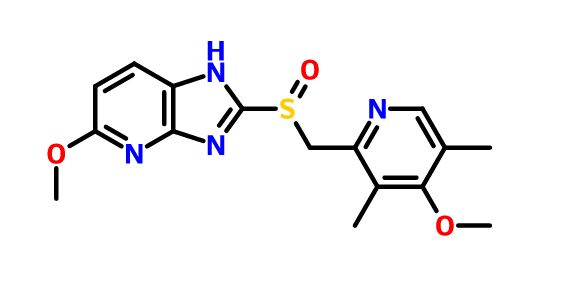
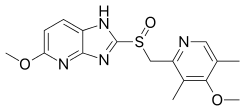
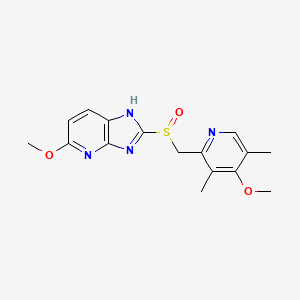
5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-imidazo[4,5-b]pyridine
2-[2-(3,5-Dimethyl)pyridylmethylsulfinyl]-5-methoxyimidazo[4,5-b]pyridine
Phase I
PHASE 1 FOR ………..A proton pump inhibitor potentially for the treatment of gastroesophageal reflux disease.
![]()
![]()
Tenatoprazole is a proton pump inhibitor drug candidate that was undergoing clinical testing as a potential treatment for refluxoesophagitis and peptic ulcer as far back as 2003.[1] The compound was invented by Mitsubishi Tanabe Pharma and was licensed to Negma Laboratories (part of Wockhardt as of 2007[2]).[3]:22
Mitsubishi reported that tenatoprazole was still in Phase I clinical trials in 2007[4]:27 and again in 2012.[3]:17
Tenatoprazole has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors, and has a half-life about seven times longer than other PPIs.[5]
![]()
Tenatoprazole is a novel imidazopyridine derivative and has an imidazopyridine ring in place of the benzimidazole moiety found in other proton pump inhibitors. It is activated more slowly than other proton pump inhibitor, but its inhibition is resistant to reversal.Tenatoprazole has an extended plasma half-life in comparison with those of all other proton pump inhibitors; this makes it more potent in the treatment of nocturnal acid breakthrough than esomeprazole, one of the most popular proton pump inhibitors.
Tenatoprazole belongs to the class of covalent proton pump inhibitors (PPIs), which is converted to the active sulfenamide or sulfenic acid by acid in the secretory canaliculus of the stimulated parietal cell of the stomach.This active species binds to luminally accessible cysteines of the gastric H+,K+-ATPase, resulting in disulfide formation and acid secretion inhibition.Tenatoprazole binds at the catalytic subunit of the gastric acid pump with a stoichiometry of 2.6 nmol mg−1 of the enzyme in vitro. In vivo, maximum binding of tenatoprazole was 2.9 nmol mg−1of the enzyme at 2 h after intravenous (IV) administration.
Tenatoprazole, or (+)-5-methoxy-2-{[(4-methoxy-3,5-dimethyl-2-pyridyl) methyl] sulfinyl} imidazo-[4,5-b] pyridine, is described in Patent No. EP 254,588. It belongs to the group of drugs considered as proton pump inhibitors, which inhibit the secretion of gastric acid and are useful in the treatment of gastric and duodenal ulcers. It can also be used to treat gastro-oesophageal reflux, digestive bleeding and dyspepsia, because of its relatively long elimination half-life, as described in the application for French patent No. FR 02. 13113.
The first known derivative of this series of proton pump inhibitors was omeprazole, described in Patent No. EP 001,529, which is endowed with properties which inhibit the secretion of gastric acid and is widely employed as an anti-ulcerative in human therapeutics.
In addition to omeprazole, other proton pump inhibitors are well known, and particular mention can be made of rabeprazole, pantoprazole and lansoprazole, which all exhibit structural analogy and lansoprazole, which all exhibit structural analogy and belong to the group of pyridinyl methyl sulfinyl benzimidazoles. These compounds are sulfoxides presenting with asymmetry at the level of the sulphur atom, and therefore generally take the form of a racemic mixture of two enantiomers.
Like omeprazole and other sulfoxide with an analogue structure, tenatoprazole has an asymmetric structure and may therefore be present in the form of a racemic mixture or of its enantiomers. Thus it may exist in the form of its two enantiomers with R and S configurations, or (+) or (−), respectively.
Recent studies have shown that, unlike all the other proton pump inhibitors such as, for example, omeprazole or lansoprazole, and unexpectedly, tenatoprazole is endowed with a markedly prolonged duration of action, resulting from a plasma half-life which is about seven times longer. Thus the clinical data collected have shown that tenatoprazole enables a degree of symptom relief and healing of gastric lesions which is superior to that achieved by other drugs belonging to the same therapeutic category of proton pump inhibitors, which thus allows its effective use in the treatment of atypical and oesophageal symptoms of gastro-oesophageal reflux, digestive bleeding and dyspepsia, as indicated above.
Studies performed by the application have made it possible to show that the two enantiomers contribute differently to the properties of tenatoprazole, and that the two enantiomers, (+) and (−) exhibit significantly different pharmacokinetic properties. Thus it is possible to prepare medicinal products with specific activity by isolating the enantiomers, and these enantiomers themselves exhibit a different pharmacokinetic profile from that of the known racemic mixture. It then becomes possible to use each of these enantiomers more effectively in precise indications for the treatment of perfectly identified pathologies.
![Tenatoprazole.png]()
Anti-ulcer drug
tenatoprazole (tenatoprazole) is a new proton pump inhibitor, by the Japanese company Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies jointly developed, has passed Phase II clinical trials. It acts on gastric parietal cells, reducing treatment of gastric ulcer, duodenal ulcer, reflux wall cell H + / K + -ATP activity, inhibition of gastric acid secretion, and H. pylori antibacterial activity, mainly for esophagitis and Zhuo – Ellison syndrome and gastric acid secretion disorders related diseases. Compared with the same types of drugs, Tenatoprazole suppress H + / K + -ATP enzyme activity is stronger, more stable, its efficacy than similar products currently widely used in clinical omeprazole strong 7 times. It has not been in the domestic market, nor ratified the production, with broad market prospects and development potential.
Proton pump inhibitors (proton pump inhibitors) for the treatment of acid-related diseases, the past ten years a wide range of clinical applications, better effect of the drug. It can quickly pass through the stomach wall membrane, gathered in a strongly acidic secretory tubules, and H + / K + -ATP enzyme (proton pump) thiol groups covalently bonded to form a disulfide bond, proton pump inactivation, inhibition of the enzyme H + / K + transport, so as to achieve the effect of acid suppression. Proton pump inhibitors and conventional clinical application of gastric acid suppression drugs H2 receptor antagonists compared with different sites of action and have different characteristics, namely acid-suppressing effect at night is good, rapid onset of acid inhibition strong and long time, easy to take these drugs can quickly and efficiently inhibit gastric acid secretion and clearance of Helicobacter pylori, it is widely used gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ellison syndrome and other diseases treatment. Currently, proton pump inhibitors has been listed on the main omeprazole, lansoprazole, pantoprazole, rabeprazole and esomeprazole.Physical and
chemical properties ofwhite or white crystalline powder, melting point 174 ~ 175 ℃. Soluble in chloroform, insoluble in alcohol and water.
This product and other proton pump inhibitors as compared to chemically stable. China had 34 omeprazole preparations from Portugal, Brazil, India, China and other 13 countries, the stability of the measurements were made. The results showed that only six products (18%) during the trial showing good physical and chemical stability of. 27 products (79%) less (including Chinese product), the active ingredient a significant chemical decomposition, color and physical properties such as dissolution, are also a corresponding change. The results of a stability test designed to compare the various proton pump inhibitors show investigated eight days at 60 ℃, relative humidity of 75%, after omeprazole decomposition only 3% of the active ingredient, the tenatoprazole 77% of the data, said Alpha pantoprazole stability far superior to omeprazole, is already developed similar products in the most promising products.
Synthesis
Matsuishi, N.; Takeda, H.; Iizumi, K.; Murakami, K.; Hisamitsu, A. US Patent 4,808,596, 1989
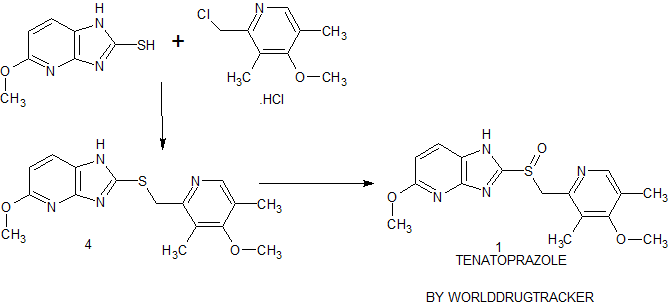
Synthesis of Tenatoprazole 1 commences with the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 in the presence of potassium hydroxide affords 4 with 73% yield in ethanol and chloroform. The oxidation of the penultimate sulfide intermediate4 with m-CPBA in chloroform (100 vol) afforded 1
![STR1]()
Syn 2
Org. Process Res. Dev., 2009, 13 (4), pp 804–806
DOI: 10.1021/op800173u
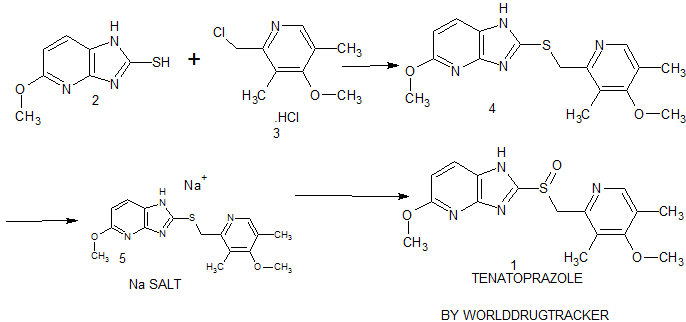
synthesis of 1 begins with the solvent-free condensation of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to deliver the sulfide intermediate4 with 98% yield.
The final step of the synthesis is the oxidation of the sulfide intermediate with m-CPBA to form tenatoprazole 1. The sulfide intermediate 4 on treatment with 0.9 equiv of m-chloroperbenzoic acid (m-CPBA) at −10 to −15 °C afforded the crude tenatoprazole which was isolated as its sodium salt. The sodium salt of tenatoprazole 5 was purified by recrystallsation using dimethyl formamide and ethyl acetate (2:1 ratio) to yield the pure crystalline tenatoprazole sodium 5. Treatment of tenatoprazole sodium 5 with dil. HCl in the presence of acetone and water afforded the pure tenatoprazole 1
![STR1]()
PATENT
CN 1861600
CN 1982311
WO 2009116072
CN 101429192
WO 2010043601
IN 2010CH00462
IN 251400
CN 102304127
WO 2012004802
CN 102703922
IN 2009DE01392
WO 2014111957
IN 2013MU00181
IN 2014CH01419
PAPER
Dai, Liyan; Synthetic Communications 2008, V38(4), P576-582
Advanced Materials Research (Durnten-Zurich, Switzerland) (2011), 233-235(Pt. 1, Fundamental of Chemical Engineering), 160-164.
Organic Process Research & Development (2013), 17(10), 1293-1299
Enantiomeric separation of proton pump inhibitors on new generation chiral columns using LC and supercritical fluid chromatography
Journal of Separation Science (2013), 36, (18), 3004-3010………http://onlinelibrary.wiley.com/doi/10.1002/jssc.201300419/abstract
PATENT
CN 102304127
https://www.google.com/patents/CN102304127A?cl=en
Tenatoprazole is a new type of gastric H + / K + -ATP enzyme inhibitors (proton pump inhibitor PPI), the chemical name 5-methoxy-2- (4-methoxy-3, 5-dimethyl-2-methylsulfinyl) imidazole and W, 5-b] pyridine, useful in the treatment of gastric ulcer, duodenal ulcer, reflux esophagitis and Zhuo – Ai syndrome and gastric acid secretion disorders related diseases. The drug was developed by Japan’s Tokyo Tanabe, Japan’s Mitsubishi Corporation and Japan’s Hokuriku pharmaceutical companies. Compared with other varieties of the same type, which inhibit H + / K + -ATP enzyme activity is stronger, ulcers of various tests are effective, and significantly improve the stability compared with other proton pump inhibitors.
US patent US4808596 “hidazo [4,5_b] pyridine compounds and pharmaceutical compositions containing same)) synthesis process disclosed Tenatoprazole the below formula:
![Figure CN102304127AD00031]()
By The route of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride with 2-mercapto-5-methoxy-imidazole, 5-b] pyridine under basic conditions condensation of Intermediate 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, and then oxidizing the Thai duly omeprazole. This route for the synthesis of pull azole classic line, many pull azoles such as omeprazole can be synthesized by a similar route, this route mild condition, simple operation. But the route condensation, oxidation treatment after use of large amounts of toxic solvent chloroform, is not conducive to industrial scale; lower oxidation yields, the resulting Tenatoprazole containing unreacted starting materials 2- [2_ (3,5 – dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine, further comprising a sulfone by-product, N- oxide, N- oxide sulfone, These by-products may interfere with purification of tenatoprazole.
Japanese Patent invention Wo 丨 J JP05222038 “5_methoxy-2- [[(4_methoxy-3, 5-dimethyl-2-pyridyl) methyl] thio] imidazo [4,5 ~ b] pyridine and intermediates)) male
Synthesis open Tenatoprazole the below formula:
4-chloro-2-chloromethyl-3,5-dimethylpyridine -N- oxide 2_ mercapto _5_ methoxy-imidazo – [4, 5-b] pyridine in alkaline under condensation of Intermediate 5-Methoxy-2- (4-chloro-3,5-dimethyl-2-methylthio Bi) imidazo W, 5-b] pyridine-oxide -N- ( yield 82%), then refluxed in a solution of sodium methoxide in methanol to give 5-methoxy-2- (4-oxo-3,5-dimethyl-2-methyl sulfide) imidazo W , 5-b] pyridine -N- oxide (income ¥ 71%), and then at room temperature in methylene chloride, phosphorus trichloride treated with deoxy (yield 95%), and finally oxidation in Tenatoprazole (income Rate not reported). The novel synthetic route, mild reaction conditions, simple operation, the yield of each step is higher, but the route is too long resulting in a total yield is not high, prolonged and rising production costs.
Reaction route is as follows:
Example 1:
] a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine:
To a reaction flask was added 2-mercapto-5-methoxy-imidazole, 5-b] pyridine 18. lg, 12g of sodium hydroxide and water 144. 8g, stirred and dissolved at 25 ° C, was added dropwise within Ih 20g of the 2-chloromethyl-dimethyl-4-methoxy _3,5- pyridine hydrochloride and 60g of water were mixed solution dropwise at 25 ° C the reaction 2h, the reaction is completed, filtered, washed with water 144. 8g, 36. 2mL ethanol and washed to obtain a wet powder; wet powder was dried at 50 ° C in vacuo to constant weight to give 2- [2_ (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5 – methoxy] imidazo [4,5-b] pyridine 32. Og;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5-b] pyridine 30g, dichloromethane 300g, methanol 15g, and dissolved with stirring; cooled to -10 ° C, was added dropwise the 15g and 485g m-chloroperbenzoic acid in methylene chloride mixed solution, dropwise addition the reaction temperature was controlled at -10 ° C, the dropping time of the pool; the dropwise addition, the temperature control at -10 ° C, the reaction 30min; completion of the reaction, at 10 ° C by the dropwise addition of lithium hydroxide and 135g water 15g mixed solution, drip complete, insulation stirred Ih; filtered cake was washed with acetone 60mL, get wet powder; wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole lithium salt ^ g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, acetone 63mL, water IOOmL, cooling M0 ° C, dropping lmol within lh / L hydrochloric pH7 0, drops. Albert, stirring 30min; the filter cake washed with water 50mL, washed with acetone and 50mL, wet powder was dried at 35 ° C under vacuum to constant weight to give Tenatoprazole 19. Sg.
Example 2:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 11. 2g of potassium hydroxide and water 217mL, stirred and dissolved at! 35 ° C, was added dropwise within 2h by the 33. 3g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 133. 2mL water mixed solution, dropwise at 35 ° C the reaction 4h, the reaction is completed, filtration, water 217mL, 72. 4mL ethanol and washed to obtain a wet powder; wet powder was dried at 60 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-pyridylmethyl sulfide -5-methoxy-yl] imidazo W, 5-b] pyridine 33. Ig;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 400mL, methanol 50mL, stirring to dissolve; cooled to _15 ° C, was added drop by the m-chloroperoxybenzoic acid 16g of mixed solution of dichloromethane and 400mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time 2. 5h; the dropwise addition, the temperature control _15 ° C, the reaction 35min; completion of the reaction, at 15 ° C by the dropwise addition of 20g of hydrogen Lithium oxide and 200mL water mixed solution, drip completed, insulation mixing 1. 5h; filtration, the filter cake washed with acetone 90mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, ethanol 75mL, water 150mL, cooled to 10 ° C, dropping 6mol / L hydrochloric pH8 0 within 2h,. drops Albert, stirring 40min; the filter cake washed with water 100mL, washed with acetone IOOmL, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 5g.
Example 3:
a) 2- [2- (3,5-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazo W, 5_b] pyridine (4) Preparation: To the reaction flask was added 2-mercapto-5-methoxy-imidazo 44,5-b] pyridine 18. lg, 8.4g of lithium hydroxide and water 180ml, stirred and dissolved at 30 ° C, was added dropwise within 1. 5h by the Guang .6g 2-chloro-3,5-dimethyl-4-methoxy-pyridine hydrochloride and 90mL water mixed solution, drop end at 30 ° C reaction 3h, the reaction is complete, filtration, water 217mL , washed with 85mL ethanol to obtain a wet powder; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5-dimethyl) -4-methoxy-5-pyridylmethyl sulfide oxy] imidazo [4,5-b] pyridine 32. 4g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 600mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 600mL , dropwise addition the reaction temperature is controlled at _20 ° C, the dropping time of the pool; the dropwise addition, the temperature control at _20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide and 300mL 30g water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 7g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, methanol 75mL, water 120mL, cooled to 5 ° C, dropping dilute hydrochloric acid within 1 5h tune pH7 5,.. drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 6g.
Example 4:
a) 2- [2- (3,5-dimethyl) -4-methoxy-picolyl thioether _5_ methoxy] imidazo [4,5_b] pyridine ⑷ Preparation of: To a solution The reaction flask was added 2-mercapto-5-methoxy imidazole -½, 5-b] pyridine 18. lg, IOg sodium hydroxide and water 150ml, stirred and dissolved at 30 ° C, the 1. 5h dropwise added from 21 . 5g of 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride and 90mL water mixed solution, dropwise at 30 ° C the reaction 3h, completion of the reaction, was filtered, washed with water 217mL, The wet powder was washed with ethanol to give 85mL; wet powder was dried at 55 ° C in vacuo to constant weight to give 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide ] imidazo [4,5-b] pyridine 32. 3g;
2) Preparation of tenatoprazole lithium salt: To a reaction flask was added 2- [2- (3,5_-dimethyl) -4-methoxy-5-methoxy-pyridylmethyl sulfide] imidazole and W, 5-b] pyridine 30g, dichloromethane 500mL, methanol 60mL, stirring to dissolve; cooled to -20 ° C, was added drop by the m-chloroperoxybenzoic acid 18g of mixed solution of dichloromethane and 500mL , the process reactor temperature control was added dropwise at -20 ° C, the dropping time pool; the dropwise addition, the temperature control in -20 ° C, the reaction 40min; completion of the reaction, at 20 ° C by the dropwise addition of lithium hydroxide 30g and 300mL water mixed solution, drip complete insulation mixing tank; filter, the filter cake washed with acetone and 120mL, get wet powder; wet powder was dried at 40 ° C under vacuum to constant weight to give Tenatoprazole lithium salt 28. 6g;
3) Preparation Tenatoprazole: To a reaction flask 加入泰 pantoprazole lithium salt 25g, isopropanol 75mL, water 120mL, cooled to 5 ° C, dropping 3mol / L hydrochloric within 1 5h. . pH7 5, drops Albert, stirring 35min; the filter cake washed with water 75mL, 75mL acetone washed, wet powder was dried at 40 ° C under vacuum to constant weight, yield powder was Tenatoprazole 19. 7g.
PAPER
An Improved Synthesis of Antiulcerative Drug: Tenatoprazole
http://pubs.acs.org/doi/full/10.1021/op800173u
Department of Research and Development, Srini Pharmaceuticals Ltd., Plot No. 10, Type-C, Road No. 8, Film Nagar, Jubilee Hills, Hyderabad-500033, Andhra Pradesh, India, Department of Chemistry, Osmania University, Tarnaka, Hyderabad-500007, Andhra Pradesh, India and Research and Development, Integrated Product Development Organization, Innovation Plaza, Dr. Reddy’s Laboratories Ltd., Bachupally, Qutubullapur, R. R. Dist. 500 072, Andhra Pradesh, India
Org. Process Res. Dev., 2009, 13 (4), pp 804–806
DOI: 10.1021/op800173u
Publication Date (Web): November 12, 2008
Copyright © 2008 American Chemical Society
* To whom correspondence should be addressed. Telephone:
+91 9490783736. E-mail:
drkvr_ou@yahoo.com;
kvgr1951@rediffmail.com., †Srini Pharmaceuticals Ltd.
, ‡Osmania University.
, §Dr. Reddy’s Laboratory Ltd.
![Abstract Image]()
An efficient, cost-effective and multikilogram-scale process for the synthesis of tenatoprazole 1, an antiulcerative drug, is described. The key steps in this synthesis involve the coupling of 2-mercapto-5-methoxyimidazo[4,5-b]pyridine 2 with 2-chloromethyl-4-methoxy-3,5-dimethyl pyridine hydrochloride 3 to yield 4 and its subsequent oxidation with m-CPBA to produce sulfoxide 1. The process has been scaled up for the multikilogram-scale of compound 1 with an overall yield of 72%. The new process requires no purification process and affords the target compound 1 with 99.8% purity by HPLC.
2-[2-(3,5-dimethyl)pyridylmethylsulfinyl]-5-methoxyimidazo[4,5-b]pyridine (1, 15.5 kg, 74%). Purity by HPLC 99.8%; 1H NMR (200 MHz, DMSO) δ 2.2 (s, 6H), 3.8 (s, 6H), 4.8 (s, 2H), 6.6 (d, 1H), 7.8 (d, 1H), 8.2 (s, 1H), 13.0 (s, 1H).
PATENT
http://www.google.co.in/patents/US7507746
the (+) enantiomer of tenatoplazole can be obtained by using chloroform, an industrially acceptable solvent, in accordance with the method proposed by Umemura et al. (J. Org. Chem. 1993, 58, 4592) as follows:
![Figure US07507746-20090324-C00001]()
Example 1 (−)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridineThe conditions for preparative chromatography, shown as an example, are as follows:
Column: 265×110 mm ChiralPak®
Chiral Stationary Phase selector of the Amylose tris type [(S)-a methylbenzylcarbamate]
Flow rate: 570 ml/min
Detection: UV 240 nm
Temperature: Ambient temperature
These conditions are implemented on a liquid preparative chromatography apparatus.
Introduce approximately 2 g of the racemic mixture if tenatoprazole exhibiting purity higher than 99.5%. The (−) enantiomer is identified by measuring the angle of optical rotation, which must be laevogyre. This measurement can be performed directly on the column, the product being dissolved in the solvent (acetonitrile).
Example 2 (+)-5-methoxy-2-{(4-methoxy-3, 5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine(R)-(+)-binaphthol 85 g (0.311 mol, 0.2 equivalence), ortho titanic acid isopropyl 42 g (0.148 mol, 0.1 equivalence), water 55 g (3.06 mol) and chloroform 7.5 L were stirred for 1 hour at room temperature. To the resultant, 5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]thio}imidazo[4,5-b]pyridine (MPI), 0.5 kg, was added and stirred for 0.5 hours at room temperature. The thus-prepared mixture was cooled to 5° C. and then 70% aqueous solution of tert-butylhydroperoxide, 0.4 L (approx. 3.0 mol, 2.0 equivalence) was added and stirred for 72 hours at the same temperature as above. After the reaction endpoint was confirmed by HPLC, an aqueous solution of sodium hydroxide was added thereto to separate the aqueous layer, thus removing foreign matter. Then, the resultant was concentrated. Ethyl acetate was added to concentrated residues, which were then heated and suspended. The thus-prepared crude crystalline substances were dissolved in water and neutralized to pH 6.8 with a diluted sulfuric acid solution which was chilled with ice. Deposited crystals were filtered, dried and recrystallized by addition of ethanol to obtain (+)-5-methoxy-2-{(4-methoxy-3,5-dimethyl-2-pyridyl)methyl]sulfinyl}-1H-imidazo[4,5-b]pyridine {(+)-TU-199}
Yield: 77%
Optical purity: 96.6% ee
Chemical purity: 94.5%
Melting point: 135° C.
Optical rotation: +184° (conditions: C=1.0, N,N-dimethylformaldehyde solution)
Ultraviolet absorption spectrum: (10 μg/mL)λmax (nm): 316, 273, 206
When measurements were carried out, for a solubility of (+), (−) forms and a racemic form (±) of tenatoprazole in relation to water, it was found that the (+) form dissolved almost 3 times greater than the racemic body and (−) form dissolved over 2 times greater than the racemic form, exhibiting favorable physical properties in preparing drugs (refer to Table 2 below).
| TABLE 2 |
| (+) form |
(−) form |
(±)racemic form |
| Solubility (water) μg/mL |
93.0 |
74.4 |
34.6 |
CLIPS
![]()
Tenatoprazole is a pyridinylmethylsulfinyl imidazopyridine compound, which is a weak base. This compound has three pKas. One is the pyridine pKa of pyridinylmethyl moiety and the others are the imidazole pKa and the pyridine pKa of the imidazopyridine moiety. The pyridine pKa1 enables tenatoprazole accumulation in the acidic canaliculus of the parietal cell. Protonation of the imidazopyridine ring enhances electron deficiency at the C-2 position, allowing intramolecular rearrangement to the active form. The active form is the sulfenic acid and/or cyclic sulfonamide, and reacts with luminal cysteine thiols of the enzyme to inhibit the enzyme activity
Synthesis route
from 2-mercapto-5-methoxy-imidazo [4,5-b] pyridine (2) and 2-chloro-3,5-dimethyl-4-methoxypyridine hydrochloride ( 3) by nucleophilic substitution synthesis of 2- (4-methoxy-3,5-dimethyl-2-methylthio) -5-methoxy-imidazo [4,5-b] pyridine (4) the oxidation of 4 1. Figure 1 is a synthesis route of tenatoprazole
![Scheme of tenatoprazole]()
References
- DataMonitor. March 2003. Gastrointestinal Disease Update: Digestive Disease Week 2003
- Economic Times. 3 March, 2011. Investors unwilling to forgive Wockhardt, promoter for failings
- Mitsubishi Tanabe Pharma State of New Product Development (as of May 8, 2012)
- Mitsubishi Tanabe Pharma FY2007 Interim Financial Results
- Li H et al. H+/K+-ATPase inhibitors: a patent review. Expert Opin Ther Pat. 2013 Jan;23(1):99-111. PMID 23205582
| US4808596 * |
24 Jul 1987 |
28 Feb 1989 |
Tokyo Tanabe Company, Ltd. |
Imidazo[4,5-b]pyridine compounds and pharmaceutical compositions containing same |
| US5753265 * |
7 Jun 1995 |
19 May 1998 |
Astra Aktiebolag |
Multiple unit pharmaceutical preparation |
| US5798120 * |
6 Oct 1994 |
25 Aug 1998 |
Tokyo Tanabe Company Limited |
Enteric granule-containing tablets |
| EP0124495A2 |
28 Feb 1984 |
7 Nov 1984 |
Aktiebolaget Hässle |
Omeprazole salts |
| EP0254588A1 |
24 Jul 1987 |
27 Jan 1988 |
Tokyo Tanabe Company Limited |
Imidazo[4,5-b] pyridine compounds, process for preparing same and pharmaceutical compositions containing same |
| Reference |
| 1 |
* |
Andersson et al., Pharmacology & Therapeutics, 2005, vol. 108, pp. 294-307. |
| 2 |
* |
Anon et al., Drugs in R&D, 2002, vol. 3, pp. 276-277. |
| 3 |
Kakinoki et al., Methods and Findings in Experimental and Clinical Pharmacology, 21(3): 179-187 (1999). |
| 4 |
Komatsu et al., J. Org. Chem., 58(17): 4529-4533 (1993). |
| 5 |
Uchiyama et al., Journal of Pharmacy and Pharmacology, 51(4): 457-464 (1999). |
| 6 |
Uchiyama et al., Methods and Findings in Experimental and Clinical Pharmacology, 21(2): 115-122 (1999). |
| Citing Patent |
Filing date |
Publication date |
Applicant |
Title |
| US20120220623 * |
30 Aug 2012 |
Mitsubishi Tanabe Pharma Corporation |
The enantiomer of tenatoprazole and the use thereof in therapy |
| CN1453278A * |
May 10, 2002 |
Nov 5, 2003 |
中国人民解放军军事医学科学院放射医学研究所 |
Omprazole compound and its prepn and application |
| CN1861600A * |
Jun 14, 2006 |
Nov 15, 2006 |
浙江大学 |
Preparation process of taytrolazole |
| Reference |
| 1 |
* |
《Organic Process Research & Development》 20081112 Somaiah Sripathi et al. An Improved Synthesis of Antiulcerative Drug:Tenatoprazole 第804-806页 1-6 第13卷, |
| 2 |
* |
《Synthetic Communication》 20080101 Liyan Dai et al. Improved Synthetic Approach to Tenatoprazole 第576-582页 1-6 第38卷, |
| 3 |
* |
《中国药物化学杂志》 20061231 赵冬梅等 抗溃疡药泰妥拉唑的合成 第360-362页 1-6 第16卷, 第6期 |
テナトプラゾール
Tenatoprazole
![]()
C16H18N4O3S : 346.4
[113712-98-4]
/////////////Tenatoprazole, 113712-98-4, TU-199, proton pump inhibitor, treatment of gastroesophageal reflux disease, Mitsubishi Tanabe Pharma, Negma Laboratories, PHASE 1, テナトプラゾール
CC1=CN=C(C(=C1OC)C)CS(=O)C2=NC3=C(N2)C=CC(=N3)OC
![Share]()















































 .
.


 ENTECAVIR, BARACLUDE
ENTECAVIR, BARACLUDE




 Rilpivirine
Rilpivirine Elbasvir
Elbasvir Quetiapine
Quetiapine























1521-3773/asset/olalertbanner.jpg?v=1&s=0c0a7bae38bad177497650f0bc0083504684f4fa)



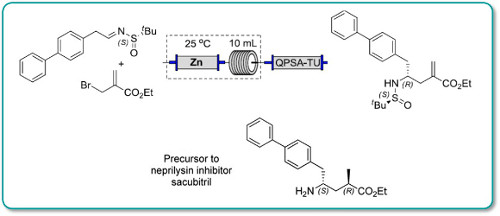






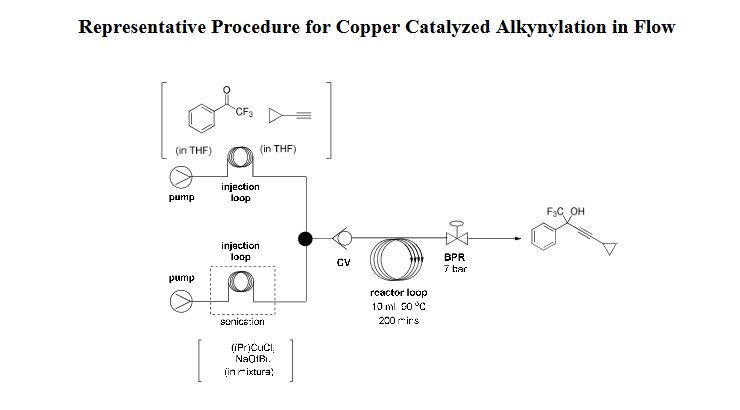
.png)


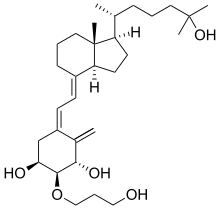
 Eldecalcitol
Eldecalcitol