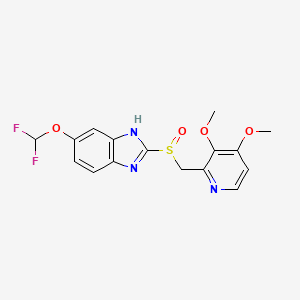
Place your arrow on above structure of Ethyl acetate………………It will flash
see label A,B,C
Integration in NMR
The intensity of the signal is proportional to the number of hydrogens that make the signal. Sometimes, NMR machines display signal intensity as an automatic display above the regular spectrum. (The exact number of hydrogens giving rise to each signal is sometimes also explicitly written above each peak, making our job a lot easier.) The intensity of the signal allows us to conclude that the more hydrogens there are in the same chemical environment, the more intense the signal will be.
Introduction
We can get the following information from a 1H Nuclear Magnetic Resonance (NMR) structure:
The number of signals gives the number of non-equivalent hydrogens
Chemical shifts show differences in the hydrogens’ chemical environments
Splitting presents the number of neighboring hydrogens (N+1 rule)
Integration gives the relative number of hydrogens present at each signal
The integrated intensity of a signal in a 1H NMR spectrum (does not apply to 13C NMR) gives a ratio for the number of hydrogens that give rise to the signal, thereby helping calculate the total number of hydrogens present in a sample.NMR machines can be used to measure signal intensity, a plot of which is sometimes automatically displayed above the regular spectrum. To show these integrations, a recorder pen marks a vertical line with a length that is proportional to the integrated area under a signal (sometimes referred to as a peak)– a value that is proportional to the number of hydrogens that are accountable for the signal. The pen then moves horizontally until another signal is reached, at which point, another vertical marking is made. We can manually measure the lengths by which the horizontal line is displaced at each peak to attain a ratio of hydrogens from the various signals. We can use this technique to figure out the hydrogen ratio when the number of hydrogens responsible for each signal is not written directly above the peak (look in the links section for an animation on how to manually find the ratio of hydrogens as described here).
Now that we’ve seen how the signal intensity is directly proportionate to the number of hydrogens that give rise to that signal, it makes sense to conclude that the more hydrogens of one kind there are in a molecule (equivalent hydrogens, so in the same chemical environment), the more intense the corresponding NMR signal will be. Here’s above a model that may help clear up some of the uncertainties.
Problems
1.) True or False? The number of hydrogens determines the intensity of a signal.
ans…………False. The relative number of hydrogens determines the intensity of a signal. The signal given by the three hydrogens in CH3CH2CHCl2 will not have the same intensity as the three hydrogens in ClCH2OCH3.
2.) Give the number of signals, the chemical shift value for each signal, and the number of integrating hydrogens for CH3OCH2CH2OCH3
answer There are 2 signals. One is at 3.3 ppm (6 hydrogens); the other at 3.5 ppm (4 hydrogens).
3
4.) answer is a and d
answer is c
Answers
False. The relative number of hydrogens determines the intensity of a signal. The signal given by the three hydrogens in CH3CH2CHCl2 will not have the same intensity as the three hydrogens in ClCH2OCH3.
There are 2 signals. One is at 3.3 ppm (6 hydrogens); the other at 3.5 ppm (4 hydrogens).
a and d
c
Number of Different Hydrogens
Ethyl acetate contains 8 hydrogens and some of them are different from each other.
For example, those labeled A are attached to a carbon bonded to a carbonyl group and are different from the hydrogens labeled B which are bonded to a carbon attached to an oxygen atom.
You can check whether certain hydrogens are the same or equivalent by replacing each hydrogen with some group X and seeing if you generate the same compound. You should convince yourself that replacing each hydrogen labeled A by X gives you identical compounds which are all equivalent by a C-C bond rotation. If this is difficult to “see” look at this molecular model of ethyl acetate to see if you can convince yourself that all the hydrogens labeled A are the same.
Integration
The area under the NMR resonance is proportional to the number of hydrogens which that resonance represents. In this way, by measuring or integrating the different NMR resonances, information regarding the relative numbers of chemically distinct hydrogens can be found. Experimentally, the integrals will appear as a line over the NMR spectrum.Integration only gives information on the relative number of different hydrogens, not the absolute number.
Review Questions
For ethyl acetate,
What ratio would you expect to see for the integrals for the hydrogens labeled A:B:C?
For ethyl ether,
What ratio would you expect to see for the integrals for the hydrogens labeled A:B?3-2
For t-butyl acetate,
What ratio would you expect to see for the integrals for the hydrogens labeled A:B:C?
ratio
2 3 3
or
4 6 6
Outside Links
Animation: how to manually find the ratio of hydrogens in a spectrum
Granulation is the act or process of forming or crystallizing into grains.[1] Granules typically have a size range between 0.2 to 4.0 mm depending on their subsequent use.
Synonym “Agglomeration”: Agglomeration processes or in a more general term particle size enlargement technologies are great tools to modify product properties. Agglomeration of powders is widely used to improve physical properties like: wettability, flowability, bulk density and product appearance.
Chemical industry
Granulation
In the chemical industry, granulation refers to the act or process in which large objects are cut or shredded and remelted into granules or pellets.
Pharmaceutical industry
In the pharmaceutical industry, granulation refers to the act or process in which primary powderparticles are made to adhere to form larger, multiparticle entities called granules. It is the process of collecting particles together by creating bonds between them. Bonds are formed by compression or by using a binding agent. Granulation is extensively used in the manufacturing of tablets and pellets (or spheroids).
The granulation process combines one or more powder particles and forms a granule that will allow tableting or spheronization process to be within required limits. This way predictable and repeatable process is possible and quality tablets or pellets can be produced using tabletting or spheronization equipment.
Purpose
Granulation is carried out for various reasons, one of those is to prevent the segregation of the constituents of powder mix. Segregation is due to differences in the size or density of the component of the mix. Normally, the smaller and/or denser particles tend to concentrate at the base of the container with the larger and/or less dense ones on the top. An ideal granulation will contain all the constituents of the mix in the correct proportion in each granule and segregation of granules will not occur.
Many powders, because of their small size, irregular shape or surface characteristics, are cohesive and do not flow well. Granules produced from such a cohesive system will be larger and more isodiametric, both factors contributing to improved flow properties.
Some powders are difficult to compact even if a readily compactable adhesive is included in the mix, but granules of the same powders are often more easily compacted. This is associated with the distribution of the adhesive within the granule and is a function of the method employed to produce the granule.
For example, if one were to make tablets from granulated sugar versus powdered sugar, powdered sugar would be difficult to compress into a tablet and granulated sugar would be easy to compress. Powdered sugar’s small particles have poor flow and compression characteristics. These small particles would have to be compressed very slowly for a long period of time to make a worthwhile tablet. Unless the powdered sugar is granulated, it could not efficiently be made into a tablet that has good tablet characteristics such as uniform content or consistent hardness.
Granulation techniques
In pharmaceutical industry, two types of granulation technologies are employed, namely, wet granulation and dry granulation.
Wet granulation
In wet granulation, granules are formed by the addition of a granulation liquid onto a powder bed which is under the influence of an impeller (in a High shear granulator, screws (in a twin screw granulator) [2] or air (in a fluidized bed granulator). The agitation resulting in the system along with the wetting of the components within the formulation results in the aggregation of the primary powder particles to produce wet granules.[2] The granulation liquid (fluid) contains a solvent which must be volatile so that it can be removed by drying, and be non-toxic. Typical liquids include water, ethanol and isopropanol either alone or in combination. The liquid solution can be either aqueous based or solvent based. Aqueous solutions have the advantage of being safer to deal with than solvents.
Water mixed into the powders can form bonds between powder particles that are strong enough to lock them together. However, once the water dries, the powders may fall apart. Therefore, water may not be strong enough to create and hold a bond. In such instances, a liquid solution that includes a binder (pharmaceutical glue) is required. Povidone, which is a polyvinyl pyrrolidone (PVP), is one of the most commonly used pharmaceutical binders. PVP is dissolved in water or solvent and added to the process. When PVP and a solvent/water are mixed with powders, PVP forms a bond with the powders during the process, and the solvent/water evaporates (dries). Once the solvent/water has been dried and the powders have formed a more densely held mass, then the granulation is milled. This process results in the formation of granules.
The process can be very simple or very complex depending on the characteristics of the powders, the final objective of tablet making, and the equipment that is available. In the traditional wet granulation method the wet mass is forced through a sieve to produce wet granules which is subsequently dried.
Dry granulation
The dry granulation process is used to form granules without using a liquid solution because the product granulated may be sensitive to moisture and heat. Forming granules without moisture requires compacting and densifying the powders. In this process the primary powder particles are aggregated under high pressure. Sweying granulator or high shear mixer-granulator can be used for the dry granulation.
Dry granulation can be conducted under two processes; either a large tablet (slug) is produced in a heavy duty tabletting press or the powder is squeezed between two counter-rotating rollers to produce a continuous sheet or ribbon of materials (roller compactor, commonly referred to as a chilsonator).
When a tablet press is used for dry granulation, the powders may not possess enough natural flow to feed the product uniformly into the die cavity, resulting in varying degrees of densification. The roller compactor (granulator-compactor) uses an auger-feed system that will consistently deliver powder uniformly between two pressure rollers. The powders are compacted into a ribbon or small pellets between these rollers and milled through a low-shear mill. When the product is compacted properly, then it can be passed through a mill and final blend before tablet compression.
^ Jump up to:ab Dhenge, Ranjit M.; Washino, Kimiaki; Cartwright, James J.; Hounslow, Michael J.; Salman, Agba D. (2012). “Twin screw granulation using conveying screws: Effects of viscosity of granulation liquids and flow of powders”. Powder Technology. doi:10.1016/j.powtec.2012.05.045.
Osborne, James; T. Althaus; L. Forny; G.Neideiretter; S.Palzer; M.Hounslow; A.D. Salman (2013). “Bonding Mechanisms Involved in the Roller Compaction of an Amorphous Material”.Chemical Engineering Science86 (5th International Granulation Workshop): 61–69. doi:10.1016/j.ces.2012.05.012.
Pharmaceutics – The science of dosage form design – M. E. Aulton 2nd EDT
Pharmaceutical dosage forms and drug delivery system – Loyd V. Allen, Nicholas G. Popovich & Howard C. Ansel 8th EDT
Lachman leon, Industrial pharmacy, special indian edition, CBS publishers
The wet granulation route of tablet manufacturing in a pharmaceutical manufacturing process is very common due to its numerous processing advantages such as enhanced powder flow and decreased segregation. However, this route is still operated in batch mode with little (if any) usage of an automatic control system. Tablet manufacturing via wet granulation, integrated with online/inline real time sensors and coupled with an automatic feedback control system, is highly desired for the transition of the pharmaceutical industry toward quality by design as opposed to quality by testing. In this manuscript, an efficient, plant-wide control strategy for an integrated continuous pharmaceutical tablet manufacturing process via wet granulation has been designed in silico. An effective controller parameter tuning strategy involving an integral of time absolute error method coupled with an optimization strategy has been used. The designed control system has been implemented in a flowsheet model that was simulated in gPROMS (Process System Enterprise) to evaluate its performance. The ability of the control system to reject the unknown disturbances and track the set point has been analyzed. Advanced techniques such as anti-windup and scale-up factor have been used to improve controller performance. Results demonstrate enhanced achievement of critical quality attributes under closed-loop operation, thus illustrating the potential of closed-loop feedback control in improving pharmaceutical tablet manufacturing operations.
Oral contraceptives, or birth control pills, have been used by more than 60 million women worldwide, and are considered by many to be the most socially significant medical advance of the twentieth century. The birth control pill is a tablet taken daily by a woman to prevent pregnancy. The birth control pill does this by inhibiting the development of the egg in the woman’s ovary during her monthly menstrual cycle. During a woman’s menstrual cycle, a low estrogen level normally triggers the pituitary gland to send out a hormone that initiates development of an egg. The birth control pill releases enough synthetic estrogen to keep that hormone from being released during the monthly cycle.
Using a process known as the wet granulation method, the active ingredients are mixed together with a dilutant and a disintegrant in a large mixer. Once mixed, the powder mass is forced through a mesh screen.
A part is pasted. article talks of manufacturing process
FDA Approves Ryanodex for the Treatment of Malignant Hyperthermia
WOODCLIFF LAKE, N.J.(BUSINESS WIRE) July 23, 2014 — Eagle Pharmaceuticals, Inc. (“Eagle” or “the Company”) (Nasdaq:EGRX) today announced that the U. S. Food and Drug Administration (FDA) has approved Ryanodex (dantrolene sodium) for injectable suspension indicated for the treatment of malignant hyperthermia (MH), along with the appropriate supportive measures. MH is an inherited and potentially fatal disorder triggered by certain anesthesia agents in genetically susceptible individuals. FDA had designated Ryanodex as an Orphan Drug in August 2013. Eagle has been informed by the FDA that it will learn over the next four to six weeks if it has been granted the seven year Orphan Drug market exclusivity.
Dantrium Intravenous is a sterile, non-pyrogenic, lyophilized formulation of dantrolene sodium for injection.
Dantrium Intravenous is supplied in 70 mL vials containing 20 mg dantrolene sodium, 3000 mg mannitol,
and sufficient sodium hydroxide to yield a pH of approximately 9.5 when reconstituted with 60 mL sterile water for injection USP (without a bacteriostatic agent).
Dantrium is classified as a direct-acting skeletal muscle relaxant. Chemically, Dantrium is hydrated 1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione sodium salt. The structural formula for the hydrated salt is:
The hydrated salt contains approximately 15% water (3-1/2 moles) and has a molecular weight of 399. The anhydrous salt (dantrolene) has a molecular weight of 336.
Dantrolene was first described in the scientific literature in 1967, as one of several hydantoin derivatives proposed as a new class of muscle relaxant.[2] Dantrolene underwent extensive further development, and its action on skeletal muscle was described in detail in 1973.[3]
Dantrolene was widely used in the management of spasticity before its efficacy in treating malignant hyperthermia was discovered by South African anesthesiologist Gaisford Harrison and reported in a landmark 1975 article published in the British Journal of Anaesthesia.[4] Harrison experimentally induced malignant hyperthermia with halothane anesthesia in genetically susceptible pigs, and obtained an 87.5% survival rate, where seven of his eight experiments survived after intravenous administration of dantrolene. The efficacy of dantrolene in humans was later confirmed in a large, multicenter study published in 1982,[5] and confirmed epidemiologically in 1993.[6] Before dantrolene, the only available treatment for malignant hyperthermia was procaine, which was associated with a 60% mortality rate in animal models.[4]
JULY 2014
The US Food and Drug Administration (FDA) has approved an injectable form of dantrolene sodium (Ryanodex, Eagle Pharmaceuticals) for rapid treatment of malignant hyperthermia (MH), along with the appropriate supportive measures, the company announced in a news release today.
MH is a potentially fatal inherited disorder triggered by exposure to certain drugs used for general anesthesia, including the neuromuscular blocking agent succinylcholine.
Ryanodex — which can be administered much more quickly than current formulations of dantrolene — is the first significant enhancement to MH treatment options in more than 3 decades, according to the company.
Ryanodex will be available in single-use vials containing 250 mg of dantrolene sodium in lyophilized powder form. It is formulated for rapid reconstitution and administration in less than 1 minute to patients in MH crisis. “Ryanodex should be administered by continuous rapid intravenous push beginning with a loading dose of 2.5 mg/kg, and continuing until symptoms subside,” the company says.
Ryanodex allows anesthesiologists to deliver a therapeutic dose of dantrolene sodium in a much more expedient manner than currently possible with existing IV formulations of dantrolene sodium, “potentially saving lives and reducing MH-related morbidity,” according to the company.
Other dantrolene sodium formulations require multiple 20-mg vials reconstituted in large volumes of sterile water, a process that can take 15 to 20 minutes to mix reconstitute and administer, the company notes.
MH during surgery is a “life-threatening emergency requiring immediate treatment including the administration of the ‘antidote’ drug dantrolene sodium,” Henry Rosenberg, MD, CPE, a founder and president of the Malignant Hyperthermia Association of the United States, said in the release.
“The ability for healthcare professionals in hospitals and surgery centers to more quickly prepare and administer this new formulation of the antidote dantrolene sodium is expected to bring the crisis under control more rapidly and prevent severe complications from MH,” he said.
The FDA granted Ryanodex orphan drug status in August 2013 and priority review status in March 2014.Ryanodex will be available to order through national and regional drug wholesalers in August with product shipping shortly after. More information is available at http://www.ryanodex.com/.
Contraindications
Oral dantrolene cannot be used by:
people with a pre-existing liver disease
people with compromised lung function
people with severe cardiovascular impairment
people with a known hypersensitivity to dantrolene
pediatric patients under five years of age
people who need good muscular balance or strength to maintain an upright position, motoric function, or proper neuromuscular balance
If the indication is a medical emergency, such as malignant hyperthermia, the only significant contraindication is hypersensitivity.
Pregnancy and breastfeeding
If needed in pregnancy, adequate human studies are lacking, therefore the drug should be given in pregnant women only if clearly indicated. It may cause hypotonia in the newborn if given closely before delivery.[1]
Dantrolene should not be given to breastfeeding mothers. If a treatment is necessary, breastfeeding should be terminated.
Adverse effects
Central nervous systemside effects are quite frequently noted and encompass speech and visual disturbances, mental depression and confusion, hallucinations, headache, insomnia and exacerbation or precipitation of seizures, and increased nervousness. Infrequent cases of respiratory depression or a feeling of suffocation have been observed. Dantrolene often causes sedation severe enough to incapacitate the patient to drive or operate machinery.
Gastrointestinal effects include bad taste, anorexia, nausea, vomiting, abdominal cramps, and diarrhea.
Hepatic side effects may be seen either as asymptomatic elevation of liver enzymes and/or bilirubin or, most severe, as fatal and nonfatal hepatitis. The risk of hepatitis is associated with the duration of treatment and the daily dose. In patients treated for hyperthermia, no liver toxicity has been observed so far.
Pleural effusion with pericarditis (oral treatment only), rare cases of bone marrow damage, diffuse myalgias, backache, dermatologic reactions, transient cardiovascular reactions, and crystalluria have additionally been seen. Muscle weakness may persist for several days following treatment.
Mutagenicity and carcinogenity
Dantrolene gave positive results in animal high dose studies (with and without enzymatic activation) regarding mutagenicity and carcinogenity. No evidence for human mutagenicity and carcinogenity has been found during the long years of clinical experience.
Skeletal formula of azumolene. The bromine atom replacing the nitro group found in dantrolene may be seen at left.
Chemically it is a hydantoin derivative, but does not exhibit antiepileptic activity like other hydantoin derivates such as phenytoin.[1]
The poor water solubility of dantrolene leads to certain difficulties in its use.[1][7] A more water-soluble analog of dantrolene, azumolene, is under development for similar indications.[7] Azumolene has a bromine residue instead of the nitro group found in dantrolene, and is 30 times more water-soluble.[1]
…………………………………………….
Bioorganic and medicinal chemistry letters, 2002 , vol. 12, 22 p. 3263 – 3265
Dantrolene sodium (1-[[5-(p-nitrophenyl) furfurylidene]-amino]hydantoin sodium salt) is described in U.S. Pat. No. 3,415,821. It is used as a skeletal muscle relaxant particularly in controlling the manifestations of clinical spasticity resulting from upper neuron disorders (Physicians’ Desk Reference, 36th Edition, 1982). It is also used in the prevention and treatment of malignant hyperthermia in humans (Friesen et al., Can. Anaesth. Soc. J. 26:319-321, 1979). In connection with the use of dantrolene sodium in hyperthermic crisis it was observed that there was an elimination of the arrhythmias accompanying such crisis [Salata et al., Effects of Dantrolene Sodium on the Electrophysiological Properties of Canine Cardiac Purkinje Fibers, J. Pharmacol. Exp. Ther. 220(1):157-166 (Jan.) 1982] incorporated herein by reference.
………………………………………….
Drug interactions
Dantrolene may interact with the following drugs:[8]
Calcium channel blockers of the diltiazem/verapamil type: Intravenous treatment with dantrolene and concomitant calcium channel blocker treatment may lead to severe cardiovascular collapse, arrhythmias, myocardial depressions, and hyperkalemia.
Snyder HR, Davis CS, Bickerton RK, Halliday RP (September 1967). “1-[(5-arylfurfurylidene)amino]hydantoins. A new class of muscle relaxants”. J Med Chem10 (5): 807–10.doi:10.1021/jm00317a011. PMID6048486.
Ellis KO, Castellion AW, Honkomp LJ, Wessels FL, Carpenter JE, Halliday RP (June 1973). “Dantrolene, a direct acting skeletal muscle relaxant”. J Pharm Sci62 (6): 948–51.doi:10.1002/jps.2600620619. PMID4712630.
Harrison GG (January 1975). “Control of the malignant hyperpyrexic syndrome in MHS swine by dantrolene sodium”. Br J Anaesth47 (1): 62–5. doi:10.1093/bja/47.1.62.PMID1148076. A reprint of the article, which became a “Citation Classic”, is available in Br J Anaesth81 (4): 626–9. PMID 9924249 (free full text).
Strazis KP, Fox AW (March 1993). “Malignant hyperthermia: review of published cases”. Anesth Analg77 (3): 297–304. doi:10.1213/00000539-199377020-00014.
Sudo RT, Carmo PL, Trachez MM, Zapata-Sudo G (March 2008). “Effects of azumolene on normal and malignant hyperthermia-susceptible skeletal muscle”. Basic Clin Pharmacol Toxicol102 (3): 308–16. doi:10.1111/j.1742-7843.2007.00156.x. PMID18047479.
Carbonyl is dad and oxygen mom hence c labelled methyl has higher chemical shift and gets a little more attention
SEE BELOW
NMR IS EASY
A chemical has Formula: C5H10O2
C5H10O2
Rule 2, omit O, gives C5H10
5 – 10/2 + 1 = 1 degree of unsaturation.
Look for 1 pi bond or aliphatic ring.
IR
The band at 1740 indicates a carbonyl, probably a saturated aliphatic ester. The bands at 3000-2850 indicate C-H alkane stretches. The bands in the region 1320-1000 could be due to C-O stretch, consistent with an ester.
The bands at 3000-2850 indicate C-H alkane stretches. The band at 3028 indicates C-H aromatic stretch; aromatics also show bands in the regions 1600-1585 and 1500-1400 (C-C in-ring stretch), and 900-675 (C-H out-of-plane). The bands in the region 1250-1020 could be due to C-N stretch. The weak, broad banc at about 3500 could be amine N-H stretch or it could be a slight contamination of an impurity (water) in the sample.
Calculate the degree of unsaturation: the answer is 5. If the degree of unsaturation is 4 or greater, look for an aromatic ring, which has a degree of unsaturation of 4 (3 double bonds plus 1 ring). In addition to an aromatic ring, the molecule can have a carbon-carbon double bond, a carbonyl, or another ring.
IR Spectrum
Look for a carbonyl, since the degree of unsaturation indicates that the compound could have a double bond and we know that the molecule has an oxygen. There is a band at 1697, suggesting an alpha, beta unsaturated aldehyde or ketone(1710-1665). To see if the compound might be an aldehyde, look for bands in the region 2830-2695. In the spectrum below, note the two bands in this region, suggesting that the compound is indeed an aldehyde.
The IR can also help determine whether or not the compound is an aromatic (although the NMR is a better diagnostic method for this). Look for the C–H stretch in aromatics from 3100-3000. There are a couple very small bands in this region.
There is one more oxygen in the molecule, it could be an ether or even an ester (if we are incorrect in assuming an aldehyde is indicated). Ether IR bands are difficult to distinguish from any other C-O stretch band – the C-O stretch of alcohols, carboxylic acids, esters, and ethers all show up in the region 1320-1000 (see Ethers).
Consult the section on Aromatics for more information on IR spectroscopy of aromatics.
NMR Spectrum
From the IR, we know that compound is probably an aromatic and an aldehyde. Aldehydes and aromatics are quite distinctive in the NMR: aldehydes show up from 9-10, usually as a small singlet; aromatic protons show up from 6.5-8.5 ppm. Let’s look at the NMR:
The singlet at 9.9 ppm indicates an aldehyde; the 4 protons from 7-8 ppm indicate a di-substituted aromatic ring.
The remaining two peaks represent an ethyl group, -CH2CH3: 2 protons split by 3 protons adjacent to 3 protons split by 2 protons. The -CH2- portion of the ethyl group is shifted downfield further than the benzylic protons this indicates that it is next to an oxygen
Let’s draw an 8-carbon 10-hydrogen molecule incorporating these features: an aldehyde, a di-substituted aromatic ring, and an ethyl group:
The molecule is drawn as the para-substituted aromatic; this is indicated by the symmetry of the peaks in the aromatic region.
The following structure with the hydrogens in different colors shows how the protons correlate with the NMR peaks:
NOTE TWO AROM -H ORTHO TO O ATOM APPEAR AT 6.9 PPM
Summary
Example is 4-ethoxybenzaldehyde:
13 C NMR OF 3-Ethoxy-4-methoxybenzaldehyde(1131-52-8)
Alittle complicated than above example
The interpretation is available in form of numbering
Rofecoxib/ˌrɒfɨˈkɒksɪb/ is a nonsteroidal anti-inflammatory drug (NSAID) that has now been withdrawn over safety concerns. It was marketed by Merck & Co. to treat osteoarthritis, acute pain conditions, and dysmenorrhoea. Rofecoxib was approved by the Food and Drug Administration (FDA) on May 20, 1999, and was marketed under the brand namesVioxx, Ceoxx, and Ceeoxx.
Rofecoxib gained widespread acceptance among physicians treating patients with arthritis and other conditions causing chronic or acute pain. Worldwide, over 80 million people were prescribed rofecoxib at some time.[1]
On September 30, 2004, Merck withdrew rofecoxib from the market because of concerns about increased risk of heart attack and stroke associated with long-term, high-dosage use. Merck withdrew the drug after disclosures that it withheld information about rofecoxib’s risks from doctors and patients for over five years, resulting in between 88,000 and 140,000 cases of serious heart disease.[2] Rofecoxib was one of the most widely used drugs ever to be withdrawn from the market. In the year before withdrawal, Merck had sales revenue of US$2.5 billion from Vioxx.[3] Merck reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens.
Rofecoxib was available on prescription in both tablet-form and as an oral suspension. It was available by injection for hospital use.
Cyclooxygenase (COX) has two well-studied isoforms, called COX-1 and COX-2. COX-1 mediates the synthesis of prostaglandinsresponsible for protection of the stomach lining, while COX-2 mediates the synthesis of prostaglandins responsible for pain and inflammation. By creating “selective” NSAIDs that inhibit COX-2, but not COX-1, the same pain relief as traditional NSAIDs is offered, but with greatly reduced risk of fatal or debilitating peptic ulcers. Rofecoxib is a selective COX-2 inhibitor, or “coxib”.
Others include Merck’s etoricoxib (Arcoxia), Pfizer’s celecoxib (Celebrex) and valdecoxib (Bextra). Interestingly, at the time of its withdrawal, rofecoxib was the only coxib with clinical evidence of its superior gastrointestinal adverse effect profile over conventional NSAIDs. This was largely based on the VIGOR (Vioxx GI Outcomes Research) study, which compared the efficacy and adverse effect profiles of rofecoxib and naproxen.[4]
Pharmacokinetics
The therapeutic recommended dosages were 12.5, 25, and 50 mg with an approximate bioavailability of 93%.[5][6][7] Rofecoxib crossed the placenta and blood–brain barrier,[5][6][8]and took 1–3 hours to reach peak plasma concentration with an effective half-life (based on steady-state levels) of approximately 17 hours.[5][7][9] The metabolic products are cis-dihydro and trans-dihydro derivatives of rofecoxib[5][9] which are primarily excreted through urine.
Fabricated efficacy studies
On March 11, 2009, Scott S. Reuben, former chief of acute pain at Baystate Medical Center, Springfield, Mass., revealed that data for 21 studies he had authored for the efficacy of the drug (along with others such as celecoxib) had been fabricated in order to augment the analgesic effects of the drugs. There is no evidence that Reuben colluded with Merck in falsifying his data. Reuben was also a former paid spokesperson for the drug company Pfizer (which owns the intellectual property rights for marketing celecoxib in the United States). The retracted studies were not submitted to either the FDA or the European Union’s regulatory agencies prior to the drug’s approval. Drug manufacturer Merckhad no comment on the disclosure.[10]
Aside from the reduced incidence of gastric ulceration, rofecoxib exhibits a similar adverse effect profile to other NSAIDs.
Prostaglandin is a large family of lipids. Prostaglandin I2/PGI2/prostacyclin is just one member of it. Prostaglandins other than PGI2 (such as PGE2) also play important roles in vascular tone regulation. Prostacyclin/thromboxane are produced by both COX-1 and COX-2, and rofecoxib suppresses just COX-2 enzyme, so there is no reason to believe that prostacyclin levels are significantly reduced by the drug. And there is no reason to believe that only the balance between quantities of prostacyclin and thromboxane is the determinant factor for vascular tone.[11] Indeed Merck has stated that there was no effect on prostacyclin production in blood vessels in animal testing.[12] Other researchers have speculated that the cardiotoxicity may be associated with maleic anhydride metabolites formed when rofecoxib becomes ionized under physiological conditions. (Reddy & Corey, 2005)
Adverse cardiovascular events
VIGOR study and publishing controversy
The VIGOR (Vioxx GI Outcomes Research) study, conducted by Bombardier, et al., which compared the efficacy and adverse effect profiles of rofecoxib and naproxen, had indicated a significant 4-fold increased risk of acute myocardial infarction (heart attack) in rofecoxib patients when compared with naproxen patients (0.4% vs 0.1%, RR 0.25) over the 12 month span of the study. The elevated risk began during the second month on rofecoxib. There was no significant difference in the mortality from cardiovascular events between the two groups, nor was there any significant difference in the rate of myocardial infarction between the rofecoxib and naproxen treatment groups in patients without high cardiovascular risk. The difference in overall risk was by the patients at higher risk of heart attack, i.e. those meeting the criteria for low-dose aspirin prophylaxis of secondary cardiovascular events (previous myocardial infarction, angina, cerebrovascular accident, transient ischemic attack, or coronary artery bypass).
Merck’s scientists interpreted the finding as a protective effect of naproxen, telling the FDA that the difference in heart attacks “is primarily due to” this protective effect (Targum, 2001). Some commentators have noted that naproxen would have to be three times as effective as aspirin to account for all of the difference (Michaels 2005), and some outside scientists warned Merck that this claim was implausible before VIGOR was published.[13] No evidence has since emerged for such a large cardioprotective effect of naproxen, although a number of studies have found protective effects similar in size to those of aspirin.[14][15] Though Dr. Topol’s 2004 paper criticized Merck’s naproxen hypothesis, he himself co-authored a 2001 JAMA article stating “because of the evidence for an antiplatelet effect of naproxen, it is difficult to assess whether the difference in cardiovascular event rates in VIGOR was due to a benefit from naproxen or to a prothrombotic effect from rofecoxib.” (Mukherjee, Nissen and Topol, 2001.)
The results of the VIGOR study were submitted to the United States Food and Drug Administration (FDA) in February 2001. In September 2001, the FDA sent a warning letter to the CEO of Merck, stating, “Your promotional campaign discounts the fact that in the VIGOR study, patients on Vioxx were observed to have a four to five fold increase in myocardial infarctions (MIs) compared to patients on the comparator non-steroidal anti-inflammatory drug (NSAID), Naprosyn (naproxen).”[16] This led to the introduction, in April 2002, of warnings on Vioxx labeling concerning the increased risk of cardiovascular events (heart attack and stroke).
Months after the preliminary version of VIGOR was published in the New England Journal of Medicine, the journal editors learned that certain data reported to the FDA were not included in the NEJM article. Several years later, when they were shown a Merck memo during the depositions for the first federal Vioxx trial, they realized that these data had been available to the authors months before publication. The editors wrote an editorial accusing the authors of deliberately withholding the data.[17] They released the editorial to the media on December 8, 2005, before giving the authors a chance to respond. NEJM editor Gregory Curfman explained that the quick release was due to the imminent presentation of his deposition testimony, which he feared would be misinterpreted in the media. He had earlier denied any relationship between the timing of the editorial and the trial. Although his testimony was not actually used in the December trial, Curfman had testified well before the publication of the editorial.[18]
The editors charged that “more than four months before the article was published, at least two of its authors were aware of critical data on an array of adverse cardiovascular events that were not included in the VIGOR article.” These additional data included three additional heart attacks, and raised the relative risk of Vioxx from 4.25-fold to 5-fold. All the additional heart attacks occurred in the group at low risk of heart attack (the “aspirin not indicated” group) and the editors noted that the omission “resulted in the misleading conclusion that there was a difference in the risk of myocardial infarction between the aspirin indicated and aspirin not indicated groups.” The relative risk for myocardial infarctions among the aspirin not indicated patients increased from 2.25 to 3 (although it remained statitistically insignificant). The editors also noted a statistically significant (2-fold) increase in risk for serious thromboembolic events for this group, an outcome that Merck had not reported in the NEJM, though it had disclosed that information publicly in March 2000, eight months before publication.[19]
The authors of the study, including the non-Merck authors, responded by claiming that the three additional heart attacks had occurred after the prespecified cutoff date for data collection and thus were appropriately not included. (Utilizing the prespecified cutoff date also meant that an additional stroke in the naproxen population was not reported.) Furthermore, they said that the additional data did not qualitatively change any of the conclusions of the study, and the results of the full analyses were disclosed to the FDA and reflected on the Vioxx warning label. They further noted that all of the data in the “omitted” table were printed in the text of the article. The authors stood by the original article.[20]
NEJM stood by its editorial, noting that the cutoff date was never mentioned in the article, nor did the authors report that the cutoff for cardiovascular adverse events was before that for gastrointestinal adverse events. The different cutoffs increased the reported benefits of Vioxx (reduced stomach problems) relative to the risks (increased heart attacks).[19]
Some scientists have accused the NEJM editorial board of making unfounded accusations.[21][22] Others have applauded the editorial. Renowned research cardiologist Eric Topol,[23] a prominent Merck critic, accused Merck of “manipulation of data” and said “I think now the scientific misconduct trial is really fully backed up”.[24] Phil Fontanarosa, executive editor of the prestigious Journal of the American Medical Association, welcomed the editorial, saying “this is another in the long list of recent examples that have generated real concerns about trust and confidence in industry-sponsored studies”.[25]
On May 15, 2006, the Wall Street Journal reported that a late night email, written by an outside public relations specialist and sent to Journal staffers hours before the Expression of Concern was released, predicted that “the rebuke would divert attention to Merck and induce the media to ignore the New England Journal of Medicine‘s own role in aiding Vioxx sales.”[26]
“Internal emails show the New England Journal’s expression of concern was timed to divert attention from a deposition in which Executive Editor Gregory Curfman made potentially damaging admissions about the journal’s handling of the Vioxx study. In the deposition, part of the Vioxx litigation, Dr. Curfman acknowledged that lax editing might have helped the authors make misleading claims in the article.” The Journal stated that NEJM‘s “ambiguous” language misled reporters into incorrectly believing that Merck had deleted data regarding the three additional heart attacks, rather than a blank table that contained no statistical information; “the New England Journal says it didn’t attempt to have these mistakes corrected.”[26]
Alzheimer’s studies
In 2000 and 2001, Merck conducted several studies of rofecoxib aimed at determining if the drug slowed the onset of Alzheimer’s disease. Merck has placed great emphasis on these studies on the grounds that they are relatively large (almost 3000 patients) and compared rofecoxib to a placebo rather than to another pain reliever. These studies found an elevated death rate among rofecoxib patients, although the deaths were not generally heart-related. However, they did not find any elevated cardiovascular risk due to rofecoxib.[27] Before 2004, Merck cited these studies as providing evidence, contrary to VIGOR, of rofecoxib’s safety.
APPROVe study
In 2001, Merck commenced the APPROVe (Adenomatous Polyp PRevention On Vioxx) study, a three-year trial with the primary aim of evaluating the efficacy of rofecoxib for theprophylaxis of colorectal polyps. Celecoxib had already been approved for this indication, and it was hoped to add this to the indications for rofecoxib as well. An additional aim of the study was to further evaluate the cardiovascular safety of rofecoxib.
The APPROVe study was terminated early when the preliminary data from the study showed an increased relative risk of adverse thrombotic cardiovascular events (includingheart attack and stroke), beginning after 18 months of rofecoxib therapy. In patients taking rofecoxib, versus placebo, the relative risk of these events was 1.92 (rofecoxib 1.50 events vs placebo 0.78 events per 100 patient years). The results from the first 18 months of the APPROVe study did not show an increased relative risk of adverse cardiovascular events. Moreover, overall and cardiovascular mortality rates were similar between the rofecoxib and placebo populations.[28]
In summary, the APPROVe study suggested that long-term use of rofecoxib resulted in nearly twice the risk of suffering a heart attack or stroke compared to patients receiving a placebo.
Other studies
Pre-approval Phase III clinical trials, like the APPROVe study, showed no increased relative risk of adverse cardiovascular events for the first eighteen months of rofecoxib usage (Merck, 2004). Others have pointed out that “study 090,” a pre-approval trial, showed a 3-fold increase in cardiovascular events compared to placebo, a 7-fold increase compared to nabumetone (another [NSAID]), and an 8-fold increase in heart attacks and strokes combined compared to both control groups.[29][30] Although this was a relatively small study and only the last result was statistically significant, critics have charged that this early finding should have prompted Merck to quickly conduct larger studies of rofecoxib’s cardiovascular safety. Merck notes that it had already begun VIGOR at the time Study 090 was completed. Although VIGOR was primarily designed to demonstrate new uses for rofecoxib, it also collected data on adverse cardiovascular outcomes.
Several very large observational studies have also found elevated risk of heart attack from rofecoxib. For example, a recent retrospective study of 113,000 elderly Canadians suggested a borderline statistically significant increased relative risk of heart attacks of 1.24 from Vioxx usage, with a relative risk of 1.73 for higher-dose Vioxx usage. (Levesque, 2005). Another study, using Kaiser Permanente data, found a 1.47 relative risk for low-dose Vioxx usage and 3.58 for high-dose Vioxx usage compared to current use of celecoxib, though the smaller number was not statistically significant, and relative risk compared to other populations was not statistically significant. (Graham, 2005).
Furthermore, a more recent meta-study of 114 randomized trials with a total of 116,000+ participants, published in JAMA, showed that Vioxx uniquely increased risk of renal (kidney) disease, and heart arrhythmia.[31]
Other COX-2 inhibitors
Any increased risk of renal and arrhythmia pathologies associated with the class of COX-2 inhibitors, e.g. celecoxib (Celebrex), valdecoxib (Bextra), parecoxib (Dynastat),lumiracoxib, and etoricoxib is not evident,[31] although smaller studies[32][33] had demonstrated such effects earlier with the use of celecoxib, valdecoxib and parecoxib.
Nevertheless, it is likely that trials of newer drugs in the category will be extended in order to supply additional evidence of cardiovascular safety. Examples are some more specific COX-2 inhibitors, including etoricoxib (Arcoxia) and lumiracoxib (Prexige), which are currently (circa 2005) undergoing Phase III/IV clinical trials.
Besides, regulatory authorities worldwide now require warnings about cardiovascular risk of COX-2 inhibitors still on the market. For example, in 2005, EU regulators required the following changes to the product information and/or packaging of all COX-2 inhibitors:[34]
Contraindications stating that COX-2 inhibitors must not be used in patients with established ischaemic heart disease and/or cerebrovascular disease (stroke), and also in patients with peripheral arterial disease
Reinforced warnings to healthcare professionals to exercise caution when prescribing COX-2 inhibitors to patients with risk factors for heart disease, such as hypertension, hyperlipidaemia (high cholesterol levels), diabetes and smoking
Given the association between cardiovascular risk and exposure to COX-2 inhibitors, doctors are advised to use the lowest effective dose for the shortest possible duration of treatment
Other NSAIDs
Since the withdrawal of Vioxx it has come to light that there may be negative cardiovascular effects with not only other COX-2 inhibitiors, but even the majority of other NSAIDs. It is only with the recent development of drugs like Vioxx that drug companies have carried out the kind of well executed trials that could establish such effects and these sort of trials have never been carried out in older “trusted” NSAIDs such as ibuprofen, diclofenac and others. The possible exceptions may be aspirin and naproxen due to their anti-platelet aggregation properties.
Withdrawal
Due to the findings of its own APPROVe study, Merck publicly announced its voluntary withdrawal of the drug from the market worldwide on September 30, 2004.[35]
In addition to its own studies, on September 23, 2004 Merck apparently received information about new research by the FDA that supported previous findings of increased risk of heart attack among rofecoxib users (Grassley, 2004). FDA analysts estimated that Vioxx caused between 88,000 and 139,000 heart attacks, 30 to 40 percent of which were probably fatal, in the five years the drug was on the market.[36]
On November 5, the medical journal The Lancet published a meta-analysis of the available studies on the safety of rofecoxib (Jüni et al., 2004). The authors concluded that, owing to the known cardiovascular risk, rofecoxib should have been withdrawn several years earlier. The Lancet published an editorial which condemned both Merck and the FDA for the continued availability of rofecoxib from 2000 until the recall. Merck responded by issuing a rebuttal of the Jüni et al. meta-analysis that noted that Jüni omitted several studies that showed no increased cardiovascular risk. (Merck & Co., 2004).
In 2005, advisory panels in both the U.S. and Canada encouraged the return of rofecoxib to the market, stating that rofecoxib’s benefits outweighed the risks for some patients. The FDA advisory panel voted 17-15 to allow the drug to return to the market despite being found to increase heart risk. The vote in Canada was 12-1, and the Canadian panel noted that the cardiovascular risks from rofecoxib seemed to be no worse than those from ibuprofen—though the panel recommended that further study was needed for all NSAIDs to fully understand their risk profiles. Notwithstanding these recommendations, Merck has not returned rofecoxib to the market.[37]
In 2005, Merck retained Debevoise & Plimpton LLP to investigate Vioxx study results and communications conducted by Merck. Through the report, it was found that Merck’s senior management acted in good faith, and that the confusion over the clinical safety of Vioxx was due to the sales team’s overzealous behavior. The report that was filed gave a timeline of the events surrounding Vioxx and showed that Merck intended to operate honestly throughout the process. Any mistakes that were made regarding the mishandling of clinical trial results and withholding of information was the result of oversight, not malicious behavior. The Martin Report did conclude that the Merck’s marketing team exaggerated the safety of Vioxx and replaced truthful information with sales tactics.[citation needed] The report was published in February 2006, and Merck was satisfied with the findings of the report and promised to consider the recommendations contained in the Martin Report. Advisers to the US Food and Drug Administration (FDA) have voted, by a narrow margin, that it should not ban Vioxx — the painkiller withdrawn by drug-maker Merck.
They also said that Pfizer’s Celebrex and Bextra, two other members of the family of painkillers known as COX-2 inhibitors, should remain available, despite the fact that they too boost patients’ risk of heart attack and stroke. url = http://www.nature.com/drugdisc/news/articles/433790b.html The recommendations of the arthritis and drug safety advisory panel offer some measure of relief to the pharmaceutical industry, which has faced a barrage of criticism for its promotion of the painkillers. But the advice of the panel, which met near Washington DC over 16–18 February, comes with several strings attached.
For example, most panel members said that manufacturers should be required to add a prominent warning about the drugs’ risks to their labels; to stop direct-to-consumer advertising of the drugs; and to include detailed, written risk information with each prescription. The panel also unanimously stated that all three painkillers “significantly increase the risk of cardiovascular events”.
The panel voted 17 to 15 against banning Vioxx (rofecoxib) entirely; the vote on Bextra (valdecoxib) was 17 to 13 with 2 abstentions; Celebrex (celecoxib) was endorsed 31 to 1. Shares of Merck, based in Whitehouse Station, New Jersey, and New York-based Pfizer closed up 13% and 7% respectively on 18 February, 2013, the day of the votes.
The FDA is expected to act on the recommendations within weeks. Although the agency usually follows the recommendations of its outside advisers, it is not bound to do so. A top official said that, in light of the closeness of some of the votes, the agency will examine the panel members’ comments in detail before deciding what to do.
An official from Merck said during the meeting that it would consider reintroducing Vioxx, which it withdrew in September 2004. On April 7, 2005, Pfizer withdrew Bextra from the U.S. market on recommendation by the FDA. Pfizer’s other painkiller, Celebrex, is still on the market.
Litigation
As of March 2006, there had been over 10,000 cases and 190 class actions filed against Merck[citation needed] over adverse cardiovascular events associated with rofecoxib and the adequacy of Merck’s warnings. The first wrongful death trial, Rogers v. Merck, was scheduled in Alabama in the spring of 2005, but was postponed after Merck argued that the plaintiff had falsified evidence of rofecoxib use.[1]
On August 19, 2005, a jury in Texas voted 10-2 to hold Merck liable for the death of Robert Ernst, a 59-year-old man who allegedly died of a rofecoxib-induced heart attack. The plaintiffs’ lead attorney was Mark Lanier. Merck argued that the death was due to cardiac arrhythmia, which had not been shown to be associated with rofecoxib use. The jury awarded Carol Ernst, widow of Robert Ernst, $253.4 million in damages. This award will almost certainly be capped at no more than US$26.1 million because of punitive damages limits under Texas law.[2] As of March 2006, the plaintiff had yet to ask the court to enter a judgment on the verdict; Merck has stated that it will appeal.
On November 3, 2005, Merck won the second case Humeston v. Merck, a personal injury case, in Atlantic City, New Jersey. The plaintiff experienced a mild myocardial infarction and claimed that rofecoxib was responsible, after having taken it for two months. Merck argued that there was no evidence that rofecoxib was the cause of Humeston’s injury and that there is no scientific evidence linking rofecoxib to cardiac events with short durations of use. The jury ruled that Merck had adequately warned doctors and patients of the drug’s risk.[3]
The first federal trial on rofecoxib, Plunkett v. Merck, began on November 29, 2005 in Houston. The trial ended in a hung jury and a mistrial was declared on December 12, 2005. According to the Wall Street Journal, the jury hung by an eight to one majority, favoring the defense. Upon retrial in February 2006 in New Orleans, where the Vioxx multidistrict litigation (MDL) is based, a jury found Merck not liable, even though the plaintiffs had the NEJM editor testify as to his objections to the VIGOR study.
On January 30, 2006, a New Jersey state court dismissed a case brought by Edgar Lee Boyd, who blamed Vioxx for gastrointestinal bleeding that he experienced after taking the drug. The judge said that Boyd failed to prove the drug caused his stomach pain and internal bleeding.
In January 2006, Garza v. Merck began trial in Rio Grande City, Texas. The plaintiff, a 71-year-old smoker with heart disease, had a fatal heart attack three weeks after finishing a one-week sample of rofecoxib. On April 21, 2006 the jury awarded the plaintiff $7 million compensatory and $25 million punitive. The Texas state court of appeals in San Antonio later rules Garza’s fatal heart attack probably resulted from pre-existing health conditions unrelated to his taking of Vioxx, thus reversing the $32 million jury award.[4]
On April 5, 2006, the jury held Merck liable for the heart attack of 77-year-old John McDarby, and awarded Mr McDarby $4.5 million in compensatory damages based on Merck’s failure to properly warn of Vioxx safety risks. After a hearing on April 11, 2006, the jury also awarded Mr McDarby an additional $9 million in punitive damages. The same jury found Merck not liable for the heart attack of 60-year-old Thomas Cona, a second plaintiff in the trial, but was liable for fraud in the sale of the drug to Cona.
Merck has reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens. Patients who claim to have suffered as a result of taking Vioxx in countries outside the US are campaigning for this to be extended.
In March 2010, an Australian class-action lawsuit against Merck ruled that Vioxx doubled the risk of heart attacks, and that Merck had breached the Trade Practices Act by selling a drug which was unfit for sale.[38]
In November 2011, Merck announced a civil settlement with the US Attorney’s Office for the District of Massachusetts, and individually with 43 US states and the District of Columbia, to resolve civil claims relating to Vioxx.[5] Under the terms of the settlement, Merck agreed to pay two-thirds of a previously recorded $950 million reserve charge in exchange for release from civil liability. Litigation with seven additional states remains outstanding. Under separate criminal proceedings, Merck plead guilty to a federal misdemeanor charge relating to the marketing of the drug across state lines, incurring a fine of $321.6 million.[6]
Other effects
Rofecoxib was shown to improve premenstrual acne vulgaris in a placebo controlled study.[39]
The oxidation of 4- (methylsulfanyl) acetophenone (X) with monoperoxyphthalic acid (MMPP) in dichloro-methane / methanol gives the corresponding sulfone (XI), which is brominated with Br2 / AlCl3 in chloroform, yielding the expected phenacyl bromide ( XII). Finally, this compound is cyclocondensed with phenylacetic acid (I) by means of 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) and triethylamine in acetonitrile. 5) Reaction of [4- (methylsulfonyl ) phenyl] phenylacetyl-ene (XIII) with CO catalyzed by Rh4 (CO) 12 in THF at 100 C in a stainless steel autoclave at 100 Atm pressure, followed by a chromatographic separation in a silicagel column to eliminate the undesired regioisomer.
……………….
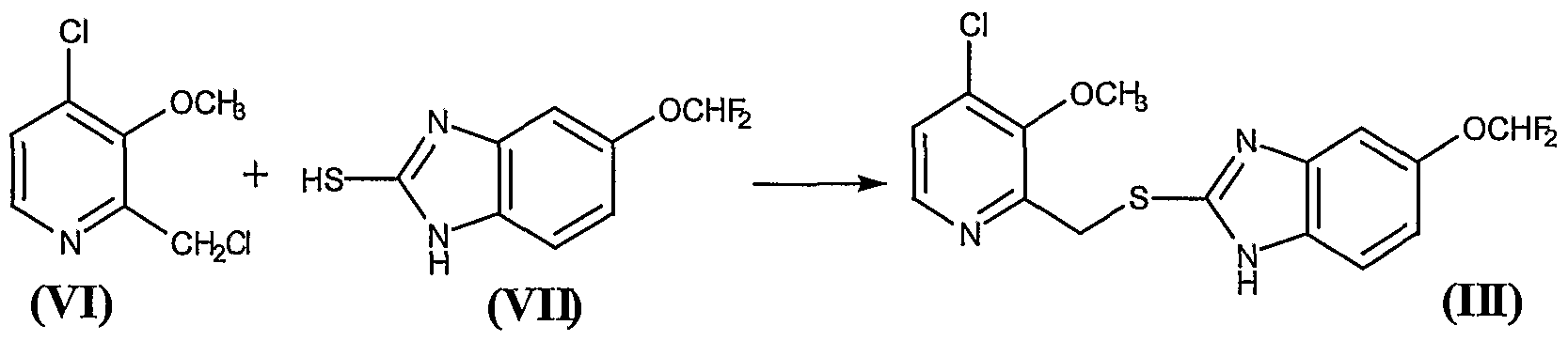
The synthesis of rofecoxib can be performed by several different ways: 1) The condensation of phenylacetic acid (I) with ethyl bromoacetate (II) by means of triethylamine in THF yields 2- (phenylacetoxy) acetic acid ethyl ester (III), which is cyclized to the hydroxyfuranone (IV) by means of potassium tert-butoxide in tert-butanol. The reaction of (IV) with triflic anhydride and diisopropylethylamine in dichloro-methane affords the corresponding triflate (V), which by reaction with LiBr in hot acetone yields the bromofuranone (VI) The condensation of (VI) with 4- (methylsulfanyl) phenylboronic acid (VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene gives 4- [4- (methylsulfanyl) -phenyl]. – 3-phenylfuran-2 (5H) -one (VIII), which is finally oxidized with 2KHSO5.KHSO4.K2SO4 (oxone). 2) The intermediate (VIII) can also be obtained by condensation of triflate (V) with boronic acid ( VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene. 3) The intermediate (VIII) can also be synthesized by the reaction of triflate (V) with tetramethylammonium chloride, giving the chlorofuranone (IX), which is then condensed with boronic acid (VII) as before.
Jump up^ Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M. B.; Hawkey, C. J.; Hochberg, M. C.; Kvien, T. K.; Schnitzer, T. J.; Vigor Study, G. (2000). “Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis”. New England Journal of Medicine343 (21): 1520–1528, 2 1528 following 1528. doi:10.1056/NEJM200011233432103. PMID11087881. edit
^ Jump up to:ab Davies, N. M.; Teng, X. W.; Skjodt, N. M. (2003). “Pharmacokinetics of rofecoxib: a specific cyclo-oxygenase-2 inhibitor”. Clinical pharmacokinetics42 (6): 545–556.PMID12793839. edit
Jump up^ Padi, S.; Kulkarni, S. (2004). “Differential effects of naproxen and rofecoxib on the development of hypersensitivity following nerve injury in rats”. Pharmacology, Biochemistry, and Behavior79 (2): 349–358. doi:10.1016/j.pbb.2004.08.005. PMID15501312. edit
Jump up^ Karha, J.; Topol, E. J. (2004). “The sad story of Vioxx, and what we should learn from it”. Cleveland Clinic journal of medicine71 (12): 933–934, 936, 934–9.doi:10.3949/ccjm.71.12.933. PMID15641522. edit
Jump up^ Solomon, D. H.; Glynn, R. J.; Levin, R.; Avorn, J. (2002). “Nonsteroidal anti-inflammatory drug use and acute myocardial infarction”. Archives of Internal Medicine162 (10): 1099–1104.doi:10.1001/archinte.162.10.1099. PMID12020178. edit
Jump up^ Curfman, G.; Morrissey, S.; Drazen, J. (2005). “Expression of concern: Bombardier et al., “Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis,” N Engl J Med 2000;343:1520-8″. The New England Journal of Medicine353 (26): 2813–2814. doi:10.1056/NEJMe058314. PMID16339408. edit
Jump up^ Bombardier, C.; Laine, L.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.; Hawkey, C.; Hochberg, M.; Kvien, T.; Schnitzer, T. J.; Weaver, A. (2006). “Response to expression of concern regarding VIGOR study”. The New England Journal of Medicine354 (11): 1196–1199. doi:10.1056/NEJMc066096. PMID16495387. edit
^ Jump up to:ab Zhang, J.; Ding, E.; Song, Y. (2006). “Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials”. Journal of the American Medical Association296 (13): 1619–1632. doi:10.1001/jama.296.13.jrv60015. PMID16968832. edit
Jump up^ Solomon, S.; McMurray, J.; Pfeffer, M.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.; Zauber, A.; Hawk, E.; Bertagnolli, M.; Adenoma Prevention with Celecoxib (APC) Study Investigators (2005). “Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention”. The New England Journal of Medicine352 (11): 1071–1080.doi:10.1056/NEJMoa050405. PMID15713944. edit
Jump up^ Nussmeier, N.; Whelton, A.; Brown, M.; Langford, R.; Hoeft, A.; Parlow, J.; Boyce, S.; Verburg, K. (2005). “Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery”. The New England Journal of Medicine352 (11): 1081–1091. doi:10.1056/NEJMoa050330. PMID15713945. edit
FDA (2005). “Summary minutes for the February 16, 17 and 18, 2005, Joint meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee.” Published on the internet, March 2005. Link
Fitzgerald GA, Coxibs and Cardiovascular Disease, N Engl J Med 2004;351(17): 1709–1711. PMID 15470192.
Merck & Co (30 Sep 2004) Merck Announces Voluntary Worldwide Withdrawal of VIOXX. Press release [7].
D. M. Mukherjee, S. E. Nissen, and E. J. Topol, “Risk of Cardiovascular Events Associated with Selective COX-2 Inhibitors,” Journal of the American Medical Association 186 (2001): 954–959.
Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 2005;352(11):1081-91. PMID 15713945
Okie, S (2005) “Raising the safety bar–the FDA’s coxib meeting.” N Engl J Med. 2005 Mar 31;352(13):1283-5. PMID 15800221.
Leleti Rajender Reddy, Corey EJ. Facile air oxidation of the conjugate base of rofecoxib (Vioxx), a possible contributor to chronic human toxicity Tetrahedron Lett 2005, 46: 927. doi:10.1016/j.tetlet.2004.12.055
Swan SK et al., Effect of Cyclooxygenase-2 Inhibition on Renal Function in Elderly Persons Receiving a Low-Salt Diet. Annals of Int Med 2000; 133:1–9
Targum, SL. (1 Feb. 2001) Review of cardiovascular safety database. FDA memorandum.[8]
Wolfe, MM et al., Gastrointestinal Toxicity of Nonsteroidal Anti-anflamattory Drugs, New England Journal of Medicine. 1999; 340; 1888-98.
Combination therapies are a promising strategy for correcting the most common gene mutation responsible for cystic fibrosis. A “corrector” drug rescues a misfolded ion channel (purple squiggle) and brings it to the cell membrane (yellow). Then, a “potentiator” drug coaxes the channel open, allowing more chloride ions (green) out.
Credit: Sci. Transl. Med.
One of the two drugs in a promising cystic fibrosis therapy might be undoing the beneficial effects of the other, two studies suggest. The work, conducted in cells, may explain the modest results from a major clinical trial and provide an avenue for improved drugs.
Identifying target product profile (TPP). TPP has been defined as a “prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and thus the safety and efficacy, of a drug product is realized”. This includes dosage form and route of administration, dosage form strength(s), therapeutic moiety release or delivery and pharmacokinetic characteristics (e.g., dissolution and aerodynamic performance) appropriate to the drug product dosage form being developed and drug product-quality criteria (e.g., sterility and purity) appropriate for the intended marketed product. The concept of TPP in this form and its application is novel in the QbD paradigm.
Identifying CQAs. Once TPP has been identified, the next step is to identify the relevant CQAs. A CQA has been defined as “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality”10. Identification of CQAs is done through risk assessment as per the ICH guidance Q9 . Prior product knowledge, such as the accumulated laboratory, nonclinical and clinical experience with a specific product-quality attribute, is key in making these risk assessments. Such knowledge may also include relevant data from similar molecules and data from literature references. Taken together, this information provides a rationale for relating the CQA to product safety and efficacy. The outcome of the risk assessment would be a list of CQAs ranked in order of importance. Use of robust risk assessment methods for identification of CQAs is novel to the QbD paradigm.
Defining product design space. After CQAs for a product have been identified, the next step is to define the product design space (that is, specifications for in-process, drug substance and drug product attributes). These specifications are established based on several sources of information that link the attributes to the safety and efficacy of the product, including, but not limited to, the following:
Clinical design space
Nonclinical studies with the product, such as binding assays, in vivo assays and in vitro cell-based assays
Clinical and nonclinical studies with similar platform products
Published literature on other similar products
Process capability with respect to the variability observed in the manufactured lots
The difference between the actual experience in the clinic and the specifications set for the product would depend on our level of understanding of the impact that the CQA under consideration can have on the safety and efficacy of the product. For example, taking host cell proteins as a CQA, it is common to propose a specification that is considerably broader than the clinical experience. This is possible because of a greater ability to use data from other platform molecules to justify the broader specifications. On the other hand, in the case of an impurity that is unique to the product, the specifications would rely solely on clinical and nonclinical studies.
In QbD, an improved understanding of the linkages between the CQA and safety and efficacy of the product is required. QbD has brought a realization of the importance of the analytical, nonclinical and animal studies in establishing these linkages and has led to the creation of novel approaches.
Defining process design space. The overall approach toward process characterization involves three key steps. First, risk analysis is performed to identify parameters for process characterization. Second, studies are designed using design of experiments (DOE), such that the data are amenable for use in understanding and defining the design space. And third, the studies are executed and the results analyzed to determine the importance of the parameters as well as their role in establishing design space.
Failure mode and effects analysis (FMEA) is commonly used to assess the potential degree of risk for every operating parameter in a systematic manner and to prioritize the activities, such as experiments, necessary to understand the impact of these parameters on overall process performance. A team consisting of representatives from process development, manufacturing and other relevant disciplines performs an assessment to determine severity, occurrence and detection. The severity score measures the seriousness of a particular failure and is based on an estimate of the severity of the potential failure effect at a local or process level and the potential failure effect at end product use or patient level. Occurrence and detection scores are based on an excursion (manufacturing deviation) outside the operating range that results in the identified failure. Although the occurrence score measures how frequently the failure might occur, the detection score indicates the probability of timely detection and correction of the excursion or the probability of detection before end product use. All three scores are multiplied to provide a risk priority number (RPN) and the RPN scores are then ranked to identify the parameters with a high enough risk to merit process characterization. FMEA outcome for a process chromatography step in a biotech process. RPN scores are calculated and operating parameters with an RPN score >50 are characterized using a qualified scaled-down model. For the case study presented here, these include gradient slope, temperature, flow rate, product loading, end of pool collection, buffer A pH, start of pool collection, volume of wash 1, buffer B pH, buffer C pH and bed height. Process characterization focused on parameters such as temperature, that have a high impact on the process (severity = 6), occur frequently in the manufacturing plant (occurrence = 6) and are difficult to quickly correct if detected (detection = 7). In contrast, parameters such as equilibration volume, with a low impact on the process (severity = 3), low occurrence (occurrence = 2) and a limited ability to detect and correct (detection = 5), were not examined in process characterization.
Cefuroxime Axetil (1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 7 2 -(Z)-(O-methyl-oxime), 1-acetate 3-carbamate.
Its molecular formula is C 20 H 22 N 4 O 10S, and it has a molecular weight of 510.48.
Cefuroxime Axetil is used orally for the treatment of patients with mild-to-moderate infections, caused by susceptible strains of the designated microorganisms.
Cefuroxime axetil is a second generation oral cephalosporinantibiotic. It was discovered by Glaxo now GlaxoSmithKline and introduced in 1987 as Zinnat.[1] It was approved by FDA on Dec 28, 1987.[2] It is available by GSK as Ceftin in US[3] and Ceftum in India.[4]
It is an acetoxyethyl ester prodrug of cefuroxime which is effective orally.[5] The activity depends on in vivohydrolysis and release of cefuroxime.
Cefuroxime is chemically (6R, 7R)-3-carbamoyloxymethyl-7-[(Z)-2-(fur-2-yl)-2-methoxy-iminoacetamido] ceph-3-em-4-carboxylic acid and has the structural Formula II:
Cefuroxime axetil having the structural Formula I:
is the 1-acetoxyethyl ester of cefuroxime, a cephalosporin antibiotic with a broad spectrum of activity against gram-positive and gram negative micro-organisms.
This compound as well as many other esters of cefuroxime, are disclosed and claimed in U.S. Pat. No. 4,267,320. According to this patent, the presence of an appropriate esterifying group, such as the 1-acetoxyethyl group of cefuroxime axetil, enhances absorption of cefuroxime from the gastrointestinal tract, whereupon the esterifying group is hydrolyzed by enzymes present in the human body.
Because of the presence of an asymmetric carbon atom at the 1-position of the 1-acetoxyethyl group, cefuroxime axetil can be produced as R and S diastereoisomers or as a racemic mixture of the R and S diastereoisomers. U.S. Pat. No. 4,267,320 discloses conventional methods for preparing a mixture of the R and S isomers in the crystalline form, as well as for separating the individual R and S diastereoisomers.
The difference in the activity of different polymorphic forms of a given drug has drawn the attention of many workers in recent years to undertake the study on polymorphism. Cefuroxime axetil is the classical example of amorphous form exhibiting higher bioavailability than the crystalline form.
U.S. Pat. No. 4,562,181 and the related U.S. Pat. Nos. 4,820,833; 4,994,567 and 5,013,833, disclose that cefuroxime axetil in amorphous form, essentially free from crystalline material and having a purity of at least 95% aside from residual solvents, has a higher bioavailability than the crystalline form while also having adequate chemical stability.
These patents disclose that highly pure cefuroxime axetil can be recovered in substantially amorphous form from a solution containing cefuroxime axetil by spray drying, roller drying, or solvent precipitation. In each case, crystalline cefuroxime axetil is dissolved in an organic solvent and the cefuroxime axetil is recovered from the solution in a highly pure, substantially amorphous form.
Another U.S. Pat. No. 5,063,224 discloses that crystalline R-cefuroxime axetil which is substantially free of S-isomer is readily absorbed from the stomach and gastrointestinal tract of animals and is therefore ideally suited to oral therapy of bacterial infections.
According to this patent, such selective administration of R-cefuroxime axetil results in surprisingly greater bioavailability ability of cefuroxime, and thus dramatically reduces the amount of unabsorbable cefuroxime remaining in the gut lumen, thereby diminishing adverse side effects attributable to cefuroxime.
British Patent Specification No. 2,145,409 discloses a process for obtaining pure crystalline cefuroxime axetil and is said to be an improvement over British Patent Specification No. 1,571,683. Sodium cefuroxime is used as the starting material in the disclosed specification, which in turn, is prepared from either 3-hydroxy cefuroxime or cefuroxime.
Said process involves an additional step of preparing sodium cefuroxime, and therefore is not economical from commercial point of view.
CEFTIN (cefuroxime axetil) Tablets and CEFTIN (cefuroxime axetil) for Oral Suspension contain cefuroxime as cefuroxime axetil. CEFTIN (cefuroxime axetil) is a semisynthetic, broad-spectrum cephalosporin antibiotic for oral administration.
Chemically, cefuroxime axetil, the 1-(acetyloxy) ethyl ester of cefuroxime, is (RS)-1-hydroxyethyl (6R,7R)-7-[2-(2-furyl)glyoxyl-amido]-3-(hydroxymethyl)-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylate, 72-(Z)-(O-methyl-oxime), 1-acetate 3-carbamate. Its molecular formula is C20H22N4O10S, and it has a molecular weight of 510.48.
Cefuroxime axetil is in the amorphous form and has the following structural formula:
CEFTIN (cefuroxime axetil) Tablets are film-coated and contain the equivalent of 250 or 500 mg of cefuroxime as cefuroxime axetil. CEFTIN (cefuroxime axetil) Tablets contain the inactive ingredients colloidal silicon dioxide, croscarmellose sodium, hydrogenated vegetable oil, hypromellose, methylparaben, microcrystalline cellulose, propylene glycol, propylparaben, sodium benzoate, sodium lauryl sulfate, and titanium dioxide.
CEFTIN (cefuroxime axetil) for Oral Suspension, when reconstituted with water, provides the equivalent of 125 mg or 250 mg of cefuroxime (as cefuroxime axetil) per 5 mL of suspension. CEFTIN (cefuroxime axetil) for Oral Suspension contains the inactive ingredients acesulfame potassium, aspartame, povidone K30, stearic acid, sucrose, tutti-frutti flavoring, and xanthan gum.
Dicyclohexylamine (17.2 g) in N,N-dimethylacetamide (50 ml) was added to a solution of cefuroxime acid (42.4 g) in N,N-dimethylacetamide (300 ml) at about −10° C. (R,S)1-Acetoxethylbromide (33.4 g) in N,N-dimethylacetamide (50 ml) was added to the above solution and the reaction mixture was stirred for 45 minutes at about −3 to 0° C. Potassium carbonate (1.1 g) was added to the reaction mixture and it was further stirred at that temperature for about 4 hours. The reaction mixture was worked up by pouring into it ethyl acetate (1.0 It), water (1.2 It) and dilute hydrochloric acid (3.5% w/w, 200 ml). The organic layer was separated and the aqueous layer was again extracted with ethyl acetate. The combined organic extracts were washed with water, dilute sodium bicarbonate solution (1%), sodium chloride solution and evaporated in vacuo to give a residue. Methanol was added to the residue and the crude product was precipitated by adding water.
The resulting precipitate was filtered off and recrystallized from the mixture of ethylacetate, methanol and hexane. The precipitated product was filtered, washed and dried to give pure crystalline cefuroxime axetil (42.5 g).
Assay (by HPLC on anhydrous basis)-98.2% w/w; Diastereoisomer ratio-0.53; Total related substances-0.48% w/w.
The present invention relates to an improved method for synthesis of cefuroxime axetil of formula (I) in high purity substantially free of the corresponding 2-cephem(Δ2)-ester of formule (II) and other impurities. The compound produced is valuable as a prodrug ester of the corresponding cephalosporin- 4-carboxylic acid derivative i. e. cefuroxime, particularly suitable for oral administration in various animal species and in man for treatment of infections caused by gram-positive and gram-negative bacteria.
BACKGROUND OF THE INVENTION
[0002]
One of the ways to improve the absorption of cephalosporin antibiotics which are poorly absorbed through the digestive tract is to prepare and administer the corresponding ester derivatives at the 4-carboxylic acid position. The esters are then readily and completely hydrolysed in vivoby enzymes present in the body to regenerate the active cephalosporin derivative having the free carboxylic acid at the 4-position.
[0003]
Among the various ester groups that can be prepared and administered only a selected few are biologically acceptable, in addition to possessing high antibacterial activity and broad antibacterial spectrum. Clinical studies on many such potential “prodrug esters” such as cefcanel daloxate (Kyoto), cefdaloxime pentexil tosilate (Hoechst Marion Roussel) and ceftrazonal bopentil (Roche), to name a few have been discontinued, while ceftizoxime alapivoxil ((Kyoto) in under Phase III clinical studies. The cephalosporin prodrug esters which have been successfully commercialised and marketed include cefcapene pivoxil (Flomox® , Shionogi), cefditoren pivoxil (Spectracef®, Meiji Seika), cefetamet pivoxil (Globocef®, Roche), cefotiam hexetil (Taketiam®, Takeda), cefpodoxime proxetil (Vantin®, Sankyo), cefteram pivoxil (Tomiron®, Toyama) and cefuroxime axetil (Ceftin® and Zinnat®, Glaxo Wellcome).
[0004]
Typically, such (3,7)-substituted-3-cephem-4-carboxylic acid esters represented by formula (I A) are synthesised by reacting the corresponding (3,7)-substituted-3- cephem-4-carboxylic acid derivative of formula (III A), with the desired haloester compound of formula (IV A) in a suitable organic solvent. The synthesis is summarised in Scheme-I, wherein in compounds of formula (I A), (II A), (III A) and (IV A) the groups R1 and R2 at the 3- and 7-positions of the β-lactam ring are substituents useful in cephalosporin chemistry ; R3 is the addendum which forms the ester function and X is halogen.
[0005]
However, the esterification reaction which essentially involves conversion of a polar acid or salt derivative to a neutral ester product invariably produces the corresponding (3,7)-substituted-2-cephem (Δ2)-4-carboxylic acid ester derivative of formula (II A) in varying amounts, arising out of isomerisation of the double bond from the 3-4 position to the 2-3 position as well as other unidentified impurities.
[0006]
It has been suggested [D. H. Bentley, et. al., Tetrahedron Lett., 1976, 41, 3739] that the isomerisation results from the ability of the 4-carboxylate anion of the starting carboxylic acid to abstract a proton from the 2-position of the 3-cephem-4-carboxylic acid ester formed, followed by reprotonation at 4-position to give the said Δ2-ester. It has also been suggested [R. B. Morin, et. al., J. Am. Chem. Soc., 1969, 91, 1401 ; R. B. Woodward, et. al., J. Am. Chem. Soc., 1966, 88, 852] that the equilibrium position for isomerisation is largely determined by the size of the ester addendum attached at the 4-carboxylic acid position.
[0007]
The 2-cephem-4-carboxylic acid esters of formula (II A) are not only unreactive as antibacterial agents but are undesired by-products. Pharmacopoeias of many countries are very stringent about the presence of the 2-cephem analogues in the finished sample of (3,7)-substituted-3-cephem-4-carboxylic acid esters and set limits for the permissible amounts of these isomers. Due to the structural similarity of the 2-cephem and 3-cephem analogues it is very difficult to separate the two isomers by conventional methods, such as chromatography as well as by fractional crystallisation. In addition to this removal of other unidentified impurities formed in the reaction, entails utilisation of tedious purification methods, thus overall resulting in,
a) considerable loss in yield, increasing the cost of manufacture and
b) a product of quality not conforming to and not easily amenable for upgradation to pharmacopoeial standards.
[0008]
Several methods are reported in the prior art for synthesis of cefuroxime axetil of formula (I) and various (3,7)-substituted-3-cephem-4-carboxylic acid esters of formula (I A), with attempts to minimise the unwanted Δ2-isomers formed in such reactions as well as conversion of the Δ2-isomer thus formed back to the desired Δ3- isomer. The prior art methods can be summarised as follows:
i) US Patent No, 4 267 320 (Gregson et. al.) describes a method for synthesis of cefuroxime axetil comprising reaction of cefuroxime acid or its alkali metal salts or onium salts with (R,S)-1-acetoxyethyl bromide in an inert organic solvent selected from N,N-dimethylacetamide, N,N-dimethylformamide, dimethyl sulfoxide, acetone, acetonitrile and hexamethylphosphoric triamide at a temperature in the range of -50 to +1150° C. The patent mentions that when alkali metal salts, specially potassium salt of cefuroxime acid are employed the reaction can be carried out in a nitrile solvent in the presence of a crown ether. When cefuroxime acid is employed the reaction is carried out in the presence of a weak inorganic base such as sodium carbonate or potassium carbonate, which is added prior to the addition of the haloester. The patent further mentions that the use of potassium carbonate in conjunction with the haloester, specially the bromo or iodo ester is preferred since it helps to minimise the formation of the Δ2-isomer. Ideally, substantially equivalent amounts of cefuroxime acid and the base is employed.
The US Patent No. 4 267 320 also describes methods, wherein the said esterification is carried out in the presence of an acid binding agent, which serve to bind hydrogen halide liberated in the reaction, thereby controlling the formation of the Δ2-isomer. The acid binding agents that are utilised include a tertiary amine base such as triethylamine or N, N-dimethylamine ; an inorganic base such as calcium carbonate or sodium bicarbonate and an oxirane compound such as ethylene oxide or propylene oxide.
However, from the examples provided in the above patent the yield of cefuroxime axetil and other (3,7)-substituted-3-cephem-4-carboxylic acid esters obtained is found to be only of about 50%, implying formation of substantial amounts of impurities in the reaction. Indeed, when cefuroxime acid is reacted with (R,S)-1-acetoxyethyl bromide in the presence of 0.55 molar equivalents of sodium carbonate or potassium carbonate in N,N-dimethylacetamide as solvent, as per the process disclosed in this patent, it is found that substantial amounts of the Δ2-isomer in a proportion ranging from 10-22% is formed, in addition to other unknown impurities. Also, substantial amounts of the starting cefuroxime acid remains unreacted even after 5 hrs of reaction. Isolation of the product generally affords a gummy material, which resists purification even after repeated crystallisations.
Moreover, the use of the acid binding agents mentioned in the above patent, specially tertiary amines and inorganic bases lead to cleavage of the β-lactam ring and also promote the undesired Δ2-isomerisation, thereby enhancing the level of impurities formed in the reaction.
ii) GB Patent No. 2 218 094 describes a method by which the Δ2-isomers formed during esterification can be converted back to the desired Δ3-isomers. The method comprises of oxidation of the dihydrothiazine ring in the mixture of Δ2- and Δ3- cephalosporin acid esters to the corresponding sulfoxide derivatives with suitable oxidising agents, whereby the Δ2-isomer gets isomerised to the corresponding Δ3-isomer during oxidation and the Δ3- cephalosporin acid ester sulfoxide is isolated. The sulfide group is regenerated back by reduction of the sulfoxide function with suitable reducing agents.
Typically, the oxidation is carried out using m-chloroperbenzoic acid and the reduction achieved by use of an alkali metal halide in presence of acetyl chloride in presence of an inert organic solvent or by use of a phosphorous trihalide.
Although, this method provides the desired Δ3-isomers in good purity, it cannot be considered as an industrially feasible method since it involves a two step process of oxidation and reduction, isolation of the intermediate products at each stage and necessary purifications, all resulting in considerable loss of the desired product and increase in the cost of manufacture. Moreover, the use of acetyl halide and phosphorous trihalide in the reduction step cannot be applied to cephalosporin derivatives that are sensitive to these reagents.
A similar method has been reported by Kaiser et. al. in J. Org. Chem., 1970, 35, 2430.
(iii)Mobasherry et. al. in J. Org. Chem., 1986, 51, 4723 describe preparation of certain Δ3-cephalosporin-4-carboxylic acid esters by reaction of the corresponding 3-cephem-4-carboxylic acids (in turn prepared form the corresponding carboxylic acid alkali metal salts) with an haloester in presence of 1.1 eq of sodium carbonate in the presence 1.2-1.5 eq of an alkyl halide and in presence of a solvent comprising of a mixture of N,N-dimethylformamide and dioxane. The authors claim that the method provides of Δ3- cephalosporin-4-carboxylic acid esters unaccompanied by the corresponding Δ2-isomer.
However, the method involves an additional step in that the starting 3-cephem-4-carboxylic acid ester derivatives are obtained from the corresponding alkali metal salts prior to reaction. In addition, longer reaction times of about 24 hrs coupled with the fact that it utilises dioxane, a potent carcinogen, not recommended by International Conference on Harmonisation (ICH) on industrial scale renders the method unattractive commercially.
Moreover, on duplication of the method exactly as described in the article it is found that about 3-4% of the corresponding Δ2-isomer is indeed formed in the reaction in addition to other unidentified impurities. Also, substantial amounts of the starting cephalosporin carboxylic acid is recovered unreacted.
(iv)Shigeto et. al. in Chem. Pharm. Bull., 1995, 43(11), 1998 have carried out the esterification of certain 7-substituted-3-cephem-4-carboxylic acid derivatives with 1-iodoethyl isopropyl carbonate in a solvent system containing a mixture of N, N-dimethylformamide and dioxane in a 3:5 ratio. A conversion to the corresponding 3-cephem- 4-carboxylate ester was achieved in only 34%, out of which the Δ2-isomer amounted to about 8%.
Esterification of 7-formamido-3-(N,N-dimethylcarbamoyloxy)methyl-3-cephem-4-carboxylic acid sodium salt with a suitable haloester in presence of solvents such as N, N-dimethylacetamide and N, N-dimethylformamide, with formation of about 0.8 to 3.0% of the Δ2-isomer is also reported in the above article by Shigeto et. al. The 7-formamido group was cleaved under acidic conditions to give the corresponding 7-amino derivative contaminated with only about 0.4% of the corresponding Δ2-isomer. The minimisation of the percentage of Δ2-isomer is attributed to the relative unstability of 7-amino-2-cephem-4-carboxylic acid esters in acidic conditions, facilitating isomerisation of the 2-cephem intermediate to the 3-cephem derivative.
However, the method does not have a general application, especially for synthesis of commercially valuable cephalosporin derivatives containing hydroxyimino or alkoxyimino substituents in the 7-amino side chain addendum, since these oxyimino functions exhibit a tendency to isomerise from the stable (Z)-configuration to the relatively undesirable(E)-configuration under acidic conditions. This would render separation of the two isomers cumbersome. Moreover, longer reaction times of about 18-20 hrs to effect the isomerisation of the double bond from the 2- position to the 3-position and use of toxic dioxane as solvent impose further limitations on the method.
(v) Demuth et. al. in J. Antibiotics, 1991, 44, 200 have utilised the N, N-dimethylformamide-dioxane system in the coupling of 1-iodocephem-4-nitrobenzyl ester with naldixic acid sodium salt and recommend use of dioxane since it reduces the basicity of the quinolone carboxylate and lowers the polarity of the reaction medium.
However, low yields of about 35% and use of toxic dioxane makes the method of little industrial application.
(vi) Wang et. al. in US Patent No. 5 498 787 claim a method for preparation of certain (3,7)-substituted-3-cephem-4-carboxylic acid prodrug esters, unaccompanied by the analogous 2-cephem esters comprising reaction of the corresponding (3,7)-substituted-3-cephem-4-carboxylic acid alkali metal salts with suitable haloesters in the presence of catalytic amounts of a quaternary ammonium or quarternary phosphonium salt. Among the prodrug esters covered in this patent is cefuroxime axetil. US Patent No. 5 498 787 claims that among the quarternary ammonium salts, such salts with acid counter ion, specially tetrabutyl ammonium sulfate (TBA+HSO4-) is the most preferred. When the molar ratio of TBA+HSO4-/cefuroxime sodium was above 0.40 no Δ2-isomer was detected, when the said molar ratio was below 0.40 and near about 0.20 the molar ratio of Δ2/Δ3 isomers formed was about 2.0%. When no TBA+HSO4- was added the molar ratio of Δ2/Δ3 isomers formed was about 10.0%. Examples 1 and 2 of this patent illustrate the esterification of cefuroxime sodium in presence of TBA+HSO4- and indicate that the Δ2-isomer was not detected after 3-12 hours of reaction. The same patent also establishes the superiority of TBA+HSO4- over other salts, specially tetrabutyl ammonium iodide (TBA+I-) since use of the latter salt resulted in considerable isomerisation of the double bond giving the undesired Δ2-isomer in predominant amounts.
The present inventors have, however, found that when cefuroxime sodium is reacted with (R,S)- 1-acetoxyethyl bromide in the presence of tetrabutylammonium sulfate (TBA+HSO4-) as per the method covered in US Patent No. 5,498 787 the same did not necessarily result in the production of the desired Δ3isomer free of the undesired Δ2 isomer and other impurities. Also, such process had limitations in that the reaction could not be completed at times even at the end of 5.0hrs. Moreover, the separation of the impurities; from the product proved cumbersome and could not be removed from the product even after successive crystallisations.
(vii) H. W. Lee et. al., Syntheic Communications, 1998, 28(23), 4345-4354 have demonstrated a method essentially similar to that claimed in US Patent No. 5 498 787 . The method of preparation of various esters of cefotaxime consists of reacting cefotaxime sodium with the requisite haloester compound in a suitable solvent and in presence of quarternary ammonium salts as phase transfer catalysts. It is claimed that when no quarternary ammonium salts are added the molar ratio (%) of Δ2/Δ3 isomers formed is about 10%. The formation of Δ2- isomer is minimised when quarternary ammonium salts are added and particularly when the molar ratio of TBA+HSO4-/cefotaxime sodium employed is 0.80 the formation of the Δ2- isomer is completely inhibited.
However, this method requires long hours (~18-24 hrs) and is carried out at higher temperatures (40-45° C) and as such may not be suitable for cephalosporin derivatives that are sensitive to heat.
(viii)H. W. Lee et. al. in Synthetic Communications, 1999, 29(11), 1873-1887 demonstrate a method for preparation of number of (3,7)-substituted-3-cephem-4-carboxylic acid esters comprising reacting the corresponding (3,7)-substituted-3-cephem-4-carboxylic acid derivatives with a base selected form cesium carbonate or cesium bicarbonate either used alone or in combination with potassium carbonate, sodium carbonate, potassium bicarbonate and sodium bicarbonate. The authors established that the formation of Δ2- isomers could be minimised by utilisation of a solvent combination ofN, N-dimethyl formamide and dioxane. The use of the latter mentioned solvent i. e. dioxane was expected to lower polarity of the reaction medium and thereby reduce the basicity of the transient 3-cephem-4-carboxylate anion formed in the reaction and thus preventing the isomerisation of the double bond from the 3-4 position to the 2-3 position.
The formation of the Δ2- isomer was found to be dependent on the amount of dioxane in the solvent mixture, the more the proportion of dioxane lesser the degree of isomerisation.
However, yields of representative esters obtained by the method are in the range of 45-85 %, implying that the reaction is accompanied by formation of substantial amounts of impurities and that the isomerisation is dependent on the nature of the substituent at 3α-position of the cephalosporin nucleus as well as on the nature of the haloester employed. Moreover, the method utilises dioxane, not desirable for reasons mentioned herein earlier and expensive cesium salts. This method, therefore, also has limited application.
(ix) Y.S. Cho et. al., in Korean J. Med. Chem., 1995, 5(1), 60-63 describe synthesis of several cephalosporin prodrug esters and their efficacy on oral administration. The esters were synthesised by reacting the corresponding cephalosporin-4-carboxylic acid derivative with the respective haloester derivative in presence of cesium carbonate and N, N-dimethylacetamide. The yields of the ester derivatives obtained are in the range of only 25-56%, indicating formation of substantial amounts of impurities in the reaction.
Example – 1
Preparation of (R, S -1-Acetoxyethyl-3-carbamoyloxymethyl-7-[(Z)-2-(fur-2-yl)-2-methoxyiminoacetamido]ceph-3-em-4-carboxylate (Cefuroxime axetil, I) :
Without use of GrouplI
/
II metal phosphate and C1-4 alcohol
[0045]
(R, S)-1-Acetoxyethyl bromide (1.6gms; 0.0094moles) was added to a mixture of cefuroxime acid (2gms; 0.0047moles) and potassium carbonate (0.326gms; 0.00235moles) in N,N-dimethylacetamide (10 ml) at 5°C and stirred at 0 to 20° C for 180 minutes Ethyl acetate was added to the reaction mixture, followed by 3% aqueous sodium bicarbonate solution (15ml). The organic layer containing the title product, Δ2 isomer (8.51%) and unidentified impurities (X1-1.86% and X2 - 3.54%) was separated and washed with 10% aqueous NaCl solution. The organic solvent was evaporated off under vacuum to give 1.08gms (44.90%) of the title compound as a gummy solid.
The reaction of 3-hydroxymethyl-7- [2- (2-furyl) -2-methoxyiminoacetamido] -3-cephem-4-carboxylic acid (I) with chlorosulfonyl isocyanate (II) and then with sodium 2-ethylhexanoate gives sodium cefuroxime (III), which is then treated with 1-bromoethyl acetate (IV) in DMA.
Why India is becoming a preferable place for foreign companies to run a pharma franchise?
Various pharma companies in abroad are looking for partners in India for their pharma franchise business. There are many reasons that influence them to show interest in India based pharma franchise industry. We all are aware of the fact that India is a place where one can easily get cheap labor and innovation. The great minds working for technology development come up with latest equipments and machineries based of better technology for production of pharma products.
Microsponges are polymeric delivery systems composed of porous microspheres. They are tiny sponge-like spherical particles with a large porous surface. Moreover, they may enhance stability, reduce side effects and modify drug release favorably. Microsponge technology has many favorable characteristics, which make it a versatile drug delivery vehicle. Microsponge Systems are based on microscopic, polymer-based microspheres that can suspend or entrap a wide variety of substances, and can then be incorporated into a formulated product such as a gel, cream, liquid or powder. The outer surface is typically porous, allowing a sustained flow of substances out of the sphere. Microsponges are porous, polymeric microspheres that are used mostly for topical use and have recently been used for oral administration. Microsponges are designed to deliver a pharmaceutical active ingredient efficiently at the minimum dose and also to enhance stability, reduce side effects, and modify drug release.
Kaity S, Maiti S, Ghosh AK, Pal D, Ghosh A, Banerjee S. Microsponges: A novel strategy for drug delivery system. J Adv Pharm Technol Res 2010;1:283-90
Kaity S, Maiti S, Ghosh AK, Pal D, Ghosh A, Banerjee S. Microsponges: A novel strategy for drug delivery system. J Adv Pharm Technol Res [serial online] 2010 [cited 2014 Aug 10];1:283-90. Available from: http://www.japtr.org/text.asp?2010/1/3/283/72416
Hefei National Laboratory for Physical Sciences at Microscale, CAS Key Laboratory of Soft Matter Chemistry & Collaborative Innovation Center of Suzhou Nano Science and Technology, University of Science and Technology of China, Hefei, P. R. China E-mail: zwang3@ustc.edu.cn; Fax: (+86) 551-360-3185
Green Chem., 2014, Advance Article
DOI: 10.1039/C4GC00932K
A catalyst-free sulfonylation of activated alkenes developed under mild conditions in water.
A catalyst-free sulfonylation reaction of activated alkenes with sulfonyl hydrazides was efficiently developed under mild and environmentally benign conditions, in water without any ligand or additive. The reaction gave a range of structurally diverse mono-substituted ethyl sulfones with excellent yields, in which the by-product was nitrogen.
The U.S. Food and Drug Administration today approved Cologuard, the first stool-based colorectal screening test that detects the presence of red blood cells and DNA mutations that may indicate the presence of certain kinds of abnormal growths that may be cancers such as colon cancer or precursors to cancer.
Colorectal cancer primarily affects people age 50 and older, and among cancers that affect both men and women, it is the third most common cancer and the second leading cause of cancer-related death in the United States, according to the Centers for Disease Control and Prevention (CDC). Colorectal cancer screening is effective at reducing illness and death related to colon cancer. The CDC estimates that if everyone age 50 or older had regular screening tests as recommended, at least 60 percent of colorectal cancer deaths could be avoided.
Colorectal cancer occurs in the colon (large intestine) or rectum (the passageway that connects the colon to the anus). Most colorectal cancers start as abnormal raised or flat tissue growths on the wall of the large intestine or rectum (polyps). Some very large polyps are called advanced adenomas and are more likely than smaller polyps to progress to cancer.
Using a stool sample, Cologuard detects hemoglobin, a protein molecule that is a component of blood. Cologuard also detects certain mutations associated with colorectal cancer in the DNA of cells shed by advanced adenomas as stool moves through the large intestine and rectum. Patients with positive test results are advised to undergo a diagnostic colonoscopy.
“This approval offers patients and physicians another option to screen for colorectal cancer,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health. “Fecal blood testing is a well-established screening tool and the clinical data showed that the test detected more cancers than a commonly used fecal occult test.”
Today’s approval of the Cologuard does not change current practice guidelines for colorectal cancer screening. Stool DNA testing (also called “fecal DNA testing”) is not currently recommended as a method to screen for colorectal cancer by the United States Preventive Services Task Force (USPSTF). Among other guidelines, the USPSTF recommends adults age 50 to 75, at average risk for colon cancer, be screened using fecal occult blood testing, sigmoidoscopy, or colonoscopy.
The safety and effectiveness of Cologuard was established in a clinical trial that screened 10,023 subjects. The trial compared the performance of Cologuard to the fecal immunochemical test (FIT), a commonly used non-invasive screening test that detects blood in the stool. Cologuard accurately detected cancers and advanced adenomas more often than the FIT test. Cologuard detected 92 percent of colorectal cancers and 42 percent of advanced adenomas in the study population, while the FIT screening test detected 74 percent of cancers and 24 percent of advanced adenomas. Cologuard was less accurate than FIT at correctly identifying subjects negative for colorectal cancer or advanced adenomas. Cologuard correctly gave a negative screening result for 87 percent of the study subjects, while FIT provided accurate negative screening results for 95 percent of the study population.
Today the Centers for Medicare & Medicaid Services (CMS) issued a proposed national coverage determination for Cologuard. Cologuard is the first product reviewed through a joint FDA-CMS pilot program known as parallel review where the agencies concurrently review medical devices to help reduce the time between the FDA’s approval of a device and Medicare coverage. This voluntary pilot program is open to certain premarket approval applications for devices with new technologies and to medical devices that fall within the scope of a Part A or Part B Medicare benefit category and have not been subject to a national coverage determination.
“Parallel review allows the last part of the FDA process to run at the same time as the CMS process, cutting as many as six months from the time from study initiation to coverage,” said Nancy Stade, CDRH’s deputy director for policy. “The pilot program is ongoing, but we will apply what we have learned to improve the efficiency of the medical device approval pathway for devices that address an important public health need.”
“This is the first time in history that FDA has approved a technology and CMS has proposed national coverage on the same day,” said Patrick Conway, chief medical officer and deputy administrator for innovation and quality for CMS. “This parallel review represents unprecedented collaboration between the two agencies and industry and most importantly will provide timely access for Medicare beneficiaries to an innovative screening test to help in the early detection of colorectal cancer.”
CMS proposes to cover the Cologuard test once every three years for Medicare beneficiaries who meet all of the following criteria:
age 50 to 85 years,
asymptomatic (no signs or symptoms of colorectal disease including but not limited to lower gastrointestinal pain, blood in stool, positive guaiac fecal occult blood test or fecal immunochemical test), and
average risk of developing colorectal cancer (no personal history of adenomatous polyps, of colorectal cancer, or inflammatory bowel disease, including Crohn’s Disease and ulcerative colitis; no family history of colorectal cancers or an adenomatous polyp, familial adenomatous polyposis, or hereditary nonpolyposis colorectal cancer).
Cologuard is manufactured by Exact Sciences in Madison, Wisconsin.
The European Medicines Agency relies on the results of clinical trials carried out by pharmaceutical companies to reach its opinions on the authorisation of medicines. Although the authorisation of clinical trials occurs at Member State level, the Agency plays a key role in ensuring that the standards of good clinical practice (GCP) are applied across the European Economic Area in cooperation with the Member States. It also manages a database of clinical trials carried out in the European Union.
Clinical trials are studies that are intended to discover or verify the effects of one or more investigational medicines. The regulation of clinical trials aims to ensure that the rights, safety and well-being of trial subjects are protected and the results of clinical trials are credible.
Regardless of where they are conducted, all clinical trials included in applications for marketing authorisation for human medicines in the European Economic Area (EEA) must have been carried out in accordance with the requirements set out in Annex 1 ofDirective 2001/83/EC. This means that:
clinical trials conducted in the EEA have to comply with European Union (EU) clinical-trial legislation (Directive 2001/20/EC);
In the EEA, approximately 4,000 clinical trials are authorised each year. This equals approximately 8,000 clinical-trial applications, with each trial involving two Member States on average. Approximately 61% of clinical trials are sponsored by the pharmaceutical industry and 39% by non-commercial sponsors, mainly academia.
Role of the Agency
Clinical-trial data is included in clinical-study reports that form a large part of the application dossiers submitted by pharmaceutical companies applying for a marketing authorisation via the Agency.
The Agency’s Committee for Medicinal Products for Human Use (CHMP) is responsible for conducting the assessment of a human medicine for which an EU-wide marketing authorisation is sought. As part of its scientific evaluation work, the CHMP reviews the clinical-trial data included in the application.
Assessments are based on purely scientific criteria and determine whether or not the medicines concerned meet the necessary quality, safety and efficacy requirements in accordance with EU legislation, particularly Directive 2001/83/EC.
Good clinical practice
The Agency plays a central role in ensuring application of good clinical practice (GCP). GCP is the international ethical and scientific quality standard for designing, recording and reporting clinical trials that involve the participation of human subjects.
The Agency works in cooperation with GCP inspectors from medicines regulatory authorities (‘national competent authorities’) in EEA Member States on the harmonisation and coordination of GCP-related activity at an EEA level.
The Agency does not have a role in the approval of clinical-trial applications in the EEA. The approval of clinical-trial applications is the responsibility of the national competent authorities.
EudraCT database and the EU Clinical Trials Register
The Agency is responsible for the development, maintenance and coordination of the EudraCT database. This is a database used by national competent authorities to enter clinical-trial data from clinical trial sponsors and paediatric-investigation-plan (PIP) addressees.
A subset of this data is made available through the European Union Clinical Trials Register, which the Agency manages on behalf of EU Member States and forms part ofEudraPharm, the EU database of medicines.
Users are able to view:
the description of phase-II to phase-IV adult clinical trials where the investigator sites are in the EEA;
the description of any clinical trials in children with investigator sites in the EU and any trials that form part of a PIP including those where the investigator sites are outside the EU.
As of 21 July 2014, it will be mandatory for sponsors to post clinical trial results in the EudraCT database. A subset of the data included in EudraCT is made available to the public in the European Union Clinical Trials Register. The content and level of detail of these summary results is set out in a European Commission guideline and in its technical guidance. A typical set of summary results provides information on the objectives of a given study, explains how it was designed and gives its main results and conclusions.
The Agency is also working towards the proactive publication of data from clinical trials carried out on the medicines that it authorises. For more information, see release of data from clinical trials.
Clinical trials conducted in countries outside the EU
Clinical trials conducted outside the EU but submitted in an application for marketing authorisation in the EU have to follow the principles which are equivalent to the provisions of the Directive 2001/20/EC.
In April 2012, the Agency published the final version of this paper:
This paper aims to strengthen existing processes to provide assurance that clinical trials meet the required ethical and GCP standards, no matter where in the world they have been conducted.
The number of clinical trials and clinical-trial subjects outside Western Europe and North America has been increasing for a number of years. More information is available in this document:
Linnaeus named the genus Aesculus after the Roman name for an edible acorn. Common names for these trees include “buckeye” and “horse chestnut”. Some are also called white chestnut or red chestnut (as in some of the Bach flower remedies). In Britain, they are sometimes called conker trees because of their link with the game of conkers, played with the seeds, also called conkers. Aesculus seeds were traditionally eaten, after leaching, by the Jōmon people of Japan over about four millennia, until 300 AD.[6]
Aesculus species have stout shoots with resinous, often sticky, buds; opposite, palmately divided leaves, often very large—to 65 cm (26 in) across in the Japanese horse chestnut Aesculus turbinata. The seeds of the Aesculus are traditionally used in a game called conkers in Europe. Species are deciduous or evergreen. Flowers are showy, insect- or bird-pollinated, with four or five petals fused into a lobedcorolla tube, arranged in a panicle inflorescence. Flowering starts after 80–110 growing degree days. The fruit matures to a capsule, 2–5 cm (25⁄32–1 31⁄32 in) diameter, usually globose, containing one to three seeds (often erroneously called a nut) per capsule. Capsules containing more than one seed result in flatness on one side of the seeds. The point of attachment of the seed in the capsule (hilum) shows as a large circular whitish scar. The capsule epidermis has “spines” (botanically: prickles) in some species, while other capsules are warty or smooth. At maturity, the capsule splits into three sections to release the seeds.[7][8][9]
The most familiar member of the genus worldwide is the common horse chestnut Aesculus hippocastanum. The yellow buckeye Aesculus flava (syn. A. octandra) is also a valuable ornamental tree with yellow flowers, but is less widely planted. Among the smaller species, the bottlebrush buckeye Aesculus parviflora also makes a very interesting and unusual flowering shrub. Several other members of the genus are used as ornamentals, and several horticultural hybrids have also been developed, most notably the red horse chestnut Aesculus × carnea, a hybrid between A. hippocastanum and A. pavia.
Use in alternative medicine
Aesculus has been listed as one of the 38 substances used to prepare Bach flower remedies,[10] a kind of alternative medicine promoted for its effect on health. However according to Cancer Research UK, “there is no scientific evidence to prove that flower remedies can control, cure or prevent any type of disease, including cancer”.[11]
Jump up^ Ogg, James G.; Gradstein, F. M; Gradstein, Felix M. (2004). A geologic time scale 2004. Cambridge, UK: Cambridge University Press.ISBN0-521-78142-6.
Jump up^ Hardin, JW. 1957. A revision of the American Hippocastanaceae I. Brittonia 9:145-171.
Jump up^ Judd, WS, RW Sanders, MJ Donoghue. 1994. Angiosperm family pairs. Harvard Papers in Botany. 1:1-51.
Jump up^ Harrington, Mark G.; Edwards, Karen J.; Johnson, Sheila A.; Chase, Mark W.; Gadek, Paul A. (Apr–Jun 2005). “Phylogenetic inference in Sapindaceae sensu lato using plastid matK and rbcL DNA sequences”. Systematic Botany30 (2): 366–382. doi:10.1600/0363644054223549. JSTOR25064067.
Jump up^ Harlan, Jack R. (1995). The Living Fields: Our Agricultural Heritage (1. publ. ed.). Cambridge [u.a.]: Cambridge Univ. Press. p. 15. ISBN0-521-40112-7.Harlan cites Akazawa, T & Aikens, CM, Prehistoric Hunter-Gathers in Japan (1986), Univ. Tokyo Press; and cites Aikens, CM & Higachi, T, Prehistory of Japan (1982), NY Academic Press.
Jump up^ Hardin, JW. 1957. A revision of the American Hippocastanaceae I. Brittonia 9:145-171
Jump up^ Hardin, JW. 1957. A revision of the American Hippocastanaceae II. Brittonia 9:173-195
Jump up^ Hardin, JW. 1960. A revision of the American Hippocastanaceae V, Species of the Old World. Brittonia 12:26-38
Forest, F., Drouin, J. N., Charest, R., Brouillet, L., & Bruneau A. (2001). A morphological phylogenetic analysis of Aesculus L. and Billia Peyr. (Sapindaceae). Canad. J. Botany79 (2): 154-169. Abstract.
A. hippocastanum grows to 36 metres (118 ft) tall, with a domed crown of stout branches; on old trees the outer branches often pendulous with curled-up tips. The leaves are opposite and palmately compound, with 5–7 leaflets; each leaflet is 13–30 cm long, making the whole leaf up to 60 cm across, with a 7–20 cm petiole. The leaf scars left on twigs after the leaves have fallen have a distinctive horseshoe shape, complete with seven “nails”. The flowers are usually white with a small red spot; they are produced in spring in erect panicles 10–30 cm tall with about 20–50 flowers on each panicle. Usually only 1–5 fruit develop on each panicle; the shell is a green, spiky capsule containing one (rarely two or three) nut-like seeds called conkers or horse-chestnuts. Each conker is 2–4 cm diameter, glossy nut-brown with a whitish scar at the base.[2]
Etymology
The common name “horse-chestnut” (often unhyphenated) is reported as having originated from the erroneous belief that the tree was a kind of chestnut (though in fact only distantly related), together with the observation that eating the fruit cured horses of chest complaints[3] despite this plant being poisonous to horses.
In Britain and Ireland, the nuts are used for the popular children’s game conkers. During the First World War, there was a campaign to ask for everyone (including children) to collect horse-chestnuts and donate them to the government. The conkers were used as a source of starch for the fermentation via the Clostridium acetobutylicum method devised by Chaim Weizmann to produce acetone. Any starch plant would have done, but they chose to ask for conkers to avoid causing starvation by using food. Weizmann’s process could use any source of starch, but it was never particularly efficient and the factory only produced acetone for three months. The aim was to produce acetone for use as solvent which aided in the production of cordite, which was then used in military armaments.
A selection of fresh conkers from a horse-chestnut
The nuts, especially those that are young and fresh, are slightly poisonous, containing alkaloidsaponins and glucosides. Although not dangerous to touch, they cause sickness when eaten; consumed by horses, they can cause tremors and lack of coordination.[6] Somemammals, notably deer, are able to break down the toxins and eat them safely.[citation needed]
Though the seeds are said to repel spiders there is little evidence to support these claims. The presence of saponin may repel insects but it is not clear whether this is effective on spiders.[7]
Horse-chestnuts have been threatened by the leaf-mining moth Cameraria ohridella, whose larvae feed on horse chestnut leaves. The moth was described from Macedonia where the species was discovered in 1984 but took 18 years to reach Britain.[8]
The flower is the symbol of the city of Kiev, capital of Ukraine.[9] Although the horse-chestnut is sometimes known as the buckeye, this name is generally reserved for the New World members of the Aesculus genus.
Medical uses
The seed extract standardized to around 20 percent aescin (escin) is used for its venotonic effect, vascular protection, anti-inflammatory and free radical scavenging properties.[10][11] Primary indication is chronic venous insufficiency.[11][12] A recent Cochrane Review found the evidence suggests that Horse Chestnut Seed Extract is an efficacious and safe short-term treatment for chronic venous insufficiency.[13]
Aescin reduces fluid leaks to surrounding tissue by reducing both the number and size of membrane pores in the veins.
Safety in medical use
Two preparations are considered; whole horsechestnut extract (whole HCE) and purified β-aescin. Historically, whole HCE has been used both for oral and IV routes (as of year 2001). The rate of adverse effects are low, in a large German study, 0.6%, consisting mainly of gastrointestinal symptoms. Dizziness, headache and itching have been reported. One serious safety issue is rare cases of acute anaphylactic reactions, presumably in a context of whole HCE. Purified β-aescin would be expected to have a better safety profile.
Another is the risk of acute renal failure, “when patients, who had undergone cardiac surgery were given high doses of horse chestnut extract i.v. for postoperative oedema. The phenomenon was dose dependent as no alteration in renal function was recorded with 340 μg kg−1, mild renal function impairment developed with 360 μg kg−1 and acute renal failure with 510 μg kg−1″.[14] This almost certainly took place in a context of whole HCE.
Three clinical trials were since performed to assess the effects of aescin on renal function. A total of 83 subjects were studied; 18 healthy volunteers given 10 or 20 mg iv. for 6 days, 40 in-patients with normal renal function given 10 mg iv. two times per day (except two children given 0.2 mg/kg), 12 patients with cerebral oedema and normal renal function given a massive iv. dose on the day of surgery (49.2 ± 19.3 mg) and 15.4 ± 9.4 mg daily for the following 10 days and 13 patients with impaired renal function due to glomerulonephritis or pyelonephritis, who were given 20–25 mg iv. daily for 6 days. “In all studies renal function was monitored daily resorting to the usual tests of renal function: BUN, serum creatinine, creatinine clearance, urinalysis. In a selected number of cases paraaminohippurate and labelled EDTA clearance were also measured. No signs of development of renal impairment in the patients with normal renal function or of worsening of renal function in the patients with renal impairment were recorded.” It is concluded that aescin has excellent tolerability in a clinical setting.[15]
Raw Horse Chestnut seed, leaf, bark and flower are toxic due to the presence of esculin and should not be ingested. Horse chestnut seed is classified by the FDA as an unsafe herb.[11] The glycoside and saponin constituents are considered toxic.[11]
Aesculus hippocastanum is used in Bach flower remedies. When the buds are used it is referred to as “chestnut bud” and when the flowers are used it is referred to as “white chestnut”.
A famous specimen of the horse-chestnut was the Anne Frank Tree in the centre of Amsterdam, which she mentioned in her diary and which survived until August 2010, when a heavy wind blew it over.[17][18] Eleven young specimens, sprouted from seeds from this tree, were transported to the United States. After a long quarantine in Indianapolis, each tree was shipped off to a new home at a notable museum or institution in the United States, such as the 9/11 Memorial Park, Central H.S. in Little Rock, and two Holocaust Centers. One of them was planted outdoors in March 2013 in front of the Children’s Museum of Indianapolis, where they were originally quarantined. [1]
Bonsai
The horse-chestnut is a favourite subject for bonsai.[19]
Diseases
Bleeding Canker. Half of all horse-chestnuts in Great Britain are now showing symptoms to some degree of this potentially lethal bacterial infection.[20][21]
Guignardia leaf blotch, caused by the fungus Guignardia aesculi
Wood rotting fungi, e.g. such as Armillaria and Ganoderma
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
Ketoconazole, 1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3– dioxolan-4-yl]methoxy]phenyl]piperazine, is a racemic mixture of the cis enantiomers (-)-(2S,4R) and (+)-(2R,4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis.(-)-Ketoconazole, the (2S,4R) enantiomer contained in the racemate of ketoconazole, is in phase III clinical trials at Cortendo for the treatment of endogenous Cushing’s syndrome. The company and licensee DiObex had also been developing the drug candidate for the treatment of type 2 diabetes; however, no recent development has been reported for this research.Preclinical studies have demonstrated the drug candidate’s ability to inhibit the synthesis of cortisol, resulting in substantial clinical benefits including lowering both blood pressure and cholesterol in addition to controlling glucose levels. It has also been shown that (-)-ketoconazole is responsible for virtually all of the cortisol synthesis inhibitory activity present in the racemate. Rights to the compound are shared with Cortendo.In 2012, orphan drug designation was assigned in the U.S. for the treatment of endogenous Cushing’s syndrome.
GÖTEBORG, Sweden.–(BUSINESS WIRE)–Cortendo AB (OSE:CORT) today announced that the first patient has been enrolled into the Phase 3 SONICS trial, i.e., “Study Of NormoCort In Cushing’s Syndrome.”
“The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”
The patient was enrolled by one of the trial’s lead principal investigators at a Pituitary Center from a prestigious institution in Baltimore, Maryland. “The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”, said Dr. Theodore R Koziol. ”The SONICS clinical trial team is acutely focused on the implementation of the trial following a successful EU Investigator’s meeting in Barcelona in July, which we believe further solidified the foundation for the trial.”
Cortendo successfully completed its European Investigator meeting supporting SONICS held in Barcelona, Spain on July 17-18. More than 35 investigators/study coordinators, including many of the world’s leading Cushing’s experts from 24 study sites, were in attendance and received training for the trial. Based on the positive feedback from the meeting, Cortendo has gained further confidence that NormoCort (COR-003) has the potential to be an important future treatment option for patients afflicted with Cushing’s Syndrome. A second US Investigator meeting is also being planned for later this year.
”It was gratifying to participate in the NormoCort SONICS trial investigator meeting in my home town of Barcelona with so many esteemed colleagues dedicated to treating patients with Cushing’s Syndrome”, said Susan Webb M.D. Ph.D. Professor of Medicine Universitat Autonoma de Barcelona. ”There remains a significant unmet medical need for patients, and I am delighted to be part of the development of this new therapy”.
Cortendo has also further strengthened its internal as well as external teams to support the study and to position the trial for an increased recruitment rate. In July, Cortendo added both an experienced physician and internal Clinical Operations Director to the NormoCort development team. Cortendo, working in concert with its CROs supporting the SONICS trial, now has a team of approximately 20 personnel on the NormoCort development program.
Cortendo has previously communicated its plan to meet the recruitment goal by increasing the number of study sites from 38 to 45 worldwide. The company is at various levels of activation with more than 30 study sites to date. Therein, Cortendo expects a large proportion of the sites to be activated by the end of the third quarter this year and remains confident that essentially all sites will be open by the end of 2014.
Risk and uncertainty
The development of pharmaceuticals carries significant risk. Failure may occur at any stage during development and commercialization due to safety or clinical efficacy issues. Delays may occur due to requirements from regulatory authorities not anticipated by the company.
About Cortendo
Cortendo AB is a biopharmaceutical company headquartered in Göteborg, Sweden. Its stock is publicly traded on the NOTC-A-list (OTC) in Norway. Cortendo is a pioneer in the field of cortisol inhibition and has completed early clinical trials in patients with Type 2 diabetes. The lead drug candidate NormoCort, the 2S, 4R-enantiomer of ketoconazole, has been re-focused to Cushing’s Syndrome, and has entered Phase 3 development. The company’s strategy is to primarily focus its resources within orphan drugs and metabolic diseases and to seek opportunities where the path to commercialization or partnership is clear and relatively near-term. Cortendo’s business model is to commercialize orphan and specialist product opportunities in key markets, and to partner non-specialist product opportunities such as diabetes at relevant development stages.
Alexander Lindström
Chief Financial Officer Office
+1 610 254 9200
Mobile : +1 917 349 7210
E-mail : alindstrom@cortendo.com
Ketoconazole, 1-acetyl-4- [4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolan-4-yl] methoxy] phenyl] piperazine, is a racemic mixture of the cis enantiomers (-)-(2S, 4R) and (+)-(2R, 4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis. Ergosterol is a key component of fungal cell walls.
More recently, ketoconazole was found to decrease plasma cortisol and to be useful, alone and in combination with other agents, in the treatment of a variety of diseases and conditions, including type 2 diabetes, Metabolic Syndrome (also known as the Insulin Resistance Syndrome, Dysmetabolic Syndrome or Syndrome X), and other medical conditions that are associated with elevated cortisol levels. SeeU.S. Patent Nos. 5,584,790 ; 6,166,017 ; and 6,642,236 , each of which is incorporated herein by reference. Cortisol is a stress-related hormone secreted from the cortex of the adrenal glands. ACTH (adenocorticotropic hormone) increases cortisol secretion. ACTH is secreted by the pituitary gland, a process activated by secretion of corticotropin releasing hormone (CRH) from the hypothalamus.
Cortisol circulates in the bloodstream and activates specific intracellular receptors, such as the glucocorticoid receptor (GR). Disturbances in cortisol levels, synthetic rates or activity have been shown to be associated with numerous metabolic complications, including insulin resistance, obesity, diabetes and Metabolic Syndrome. Additionally, these metabolic abnormalities are associated with substantially increased risk of cardiovascular disease, a major cause of death in industrialized countries. See Mårin P et al., “Cortisol secretion in relation to body fat distribution in obese premenopausal women.” Metabolism 1992; 41:882-886, Bjorntorp, “Neuroendocrine perturbations as a cause of insulin resistance.” Diabetes Metab Res Rev 1999; 15(6): 427-41, and Rosmond, “Role of stress in the pathogenesis of the metabolic syndrome.” Psychoneuroendocrinology 2005; 30(1): 1-10, each of which is incorporated herein by reference.
While ketoconazole is known to inhibit some of the enzymatic steps in cortisol synthesis, such as, for example, 17α hydroxylase (Wachall et al., “Imidazole substituted biphenyls: a new class of highly potent and in vivo active inhibitors of P450 17 as potential therapeutics for treatment of prostate cancer.” Bioorg Med Chem 1999; 7(9): 1913-24, incorporated herein by reference) and 11b-hydroxylase (Rotstein et al., “Stereoisomers of ketoconazole: preparation and biological activity.” J Med Chem 1992; 35(15): 2818-25) and 11β-hydroxy steroid dehydrogenase (11β-HSD) (Diederich et al., “In the search for specific inhibitors of human 11β-hydroxysteroid-dehydrogenases (11β-HSDs): chenodeoxycholic acid selectively inhibits 11β-HSD-L” Eur J Endocrinol 2000; 142(2): 200-7, incorporated herein by reference) the mechanisms by which ketoconazole decreases cortisol levels in the plasma have not been reported. For example, there is uncertainty regarding the effect of ketoconazole on the 11β-hydroxy steroid dehydrogenase (11β-HSD) enzymes. There are two 11β-HSD enzymes. One of these, 11β-HSD-I, is primarily a reductase that is highly expressed in the liver and can convert the inactive 11-keto glucocorticoid to the active glucocorticoid (cortisol in humans and corticosterone in rats). In contrast, the other, 11β-HSD-II, is primarily expressed in the kidney and acts primarily as an oxidase that converts active glucocorticoid (cortisol in humans and corticosterone in rats) to inactive 11-keto glucocorticoids. Thus, the plasma concentration of active glucocorticoid is influenced by the rate of synthesis, controlled in part by the activity of adrenal 11β-hydroxylase and by the rate of interconversion, controlled in part by the relative activities of the two 11β-HSD enzymes. Ketoconazole is known to inhibit these three enzymes (Diederich et al., supra) and the 2S,4R enantiomer is more active against the adrenal 11β-hydroxylase enzyme than is the 2R,4S enantiomer (Rotstein et al., supra). However, there are no reports describing the effect of the two ketoconazole enantiomers on either of 11β-HSD-I or 11β-HSD-II, so it is not possible to predict what effects, if any, the two different ketoconazole enantiomers will each have on plasma levels of the active glucocorticoid levels in a mammal.
Ketoconazole has also been reported to lower cholesterol levels in humans (Sonino et al. (1991). “Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients.” Clin Endocrinol (Oxf). 35(4): 347-52; Gylling et al. (1993). “Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia.” J Lipid Res. 34(1): 59-67) each of which is incorporated herein by reference). The 2S,4R enantiomer is more active against the cholesterol synthetic enzyme 14 αlanosterol demethylase than is the other (2R,4S) enantiomer (Rotstein et al infra). However, because cholesterol level in a human patient is controlled by the rate of metabolism and excretion as well as by the rate of synthesis it is not possible to predict from this whether the 2S,4R enantiomer of ketoconazole will be more effective at lowering cholesterol levels.
The use of ketoconazole as a therapeutic is complicated by the effect of ketoconazole on the P450 enzymes responsible for drug metabolism. Several of these P450 enzymes are inhibited by ketoconazole (Rotsteinet al., supra). This inhibition leads to an alteration in the clearance of ketoconazole itself (Brass et al., “Disposition of ketoconazole, an oral antifungal, in humans.” Antimicrob Agents Chemother 1982; 21(1): 151-8, incorporated herein by reference) and several other important drugs such as Glivec (Dutreix et al., “Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects.” Cancer Chemother Pharmacol 2004; 54(4): 290-4) and methylprednisolone (Glynn et al., “Effects of ketoconazole on methylprednisolone pharmacokinetics and cortisol secretion.” Clin Pharmacol Ther 1986; 39(6): 654-9). As a result, the exposure of a patient to ketoconazole increases with repeated dosing, despite no increase in the amount of drug administered to the patient. This exposure and increase in exposure can be measured and demonstrated using the “Area under the Curve” (AUC) or the product of the concentration of the drug found in the plasma and the time period over which the measurements are made. The AUC for ketoconazole following the first exposure is significantly less than the AUC for ketoconazole after repeated exposures. This increase in drug exposure means that it is difficult to provide an accurate and consistent dose of the drug to a patient. Further, the increase in drug exposure increases the likelihood of adverse side effects associated with ketoconazole use.
[0008]
Rotstein et al. (Rotstein et al., supra) have examined the effects of the two ketoconazole cis enantiomers on the principal P450 enzymes responsible for drug metabolism and reported “…almost no selectivity was observed for the ketoconazole isomers” and, referring to drug metabolizing P450 enzymes: “[t]he IC50 values for the cis enantiomers were similar to those previously reported for racemic ketoconazole”. This report indicated that both of the cis enantiomers could contribute significantly to the AUC problem observed with the ketoconazole racemate.
One of the adverse side effects of ketoconazole administration exacerbated by this AUC problem is liver reactions. Asymptomatic liver reactions can be measured by an increase in the level of liver specific enzymes found in the serum and an increase in these enzymes has been noted in ketoconazole treated patients (Sohn, “Evaluation of ketoconazole.” Clin Pharm 1982; 1(3): 217-24, and Janssen and Symoens, “Hepatic reactions during ketoconazole treatment.” Am J Med 1983; 74(1B): 80-5, each of which is incorporated herein by reference). In addition 1:12,000 patients will have more severe liver failure (Smith and Henry, “Ketoconazole: an orally effective antifungal agent. Mechanism of action, pharmacology, clinical efficacy and adverse effects.” Pharmacotherapy 1984; 4(4): 199-204, incorporated herein by reference). As noted above, the amount of ketoconazole that a patient is exposed to increases with repeated dosing even though the amount of drug taken per day does not increase (the “AUC problem”). The AUC correlates with liver damage in rabbits (Ma et al., “Hepatotoxicity and toxicokinetics of ketoconazole in rabbits.” Acta Pharmacol Sin 2003; 24(8): 778-782 incorporated herein by reference) and increased exposure to the drug is believed to increase the frequency of liver damage reported in ketoconazole treated patients.
Additionally, U.S. Patent No. 6,040,307 , incorporated herein by reference, reports that the 2S,4R enantiomer is efficacious in treating fungal infections. This same patent application also reports studies on isolated guinea pig hearts that show that the administration of racemic ketoconazole may be associated with an increased risk of cardiac arrhythmia, but provides no data in support of that assertion. However, as disclosed in that patent, arrhythmia had not been previously reported as a side effect of systemic racemic ketoconazole, although a particular subtype of arrhythmia, torsades de pointes, has been reported when racemic ketoconazole was administered concurrently with terfenadine. Furthermore several published reports (for example, Morganroth et al. (1997). “Lack of effect of azelastine and ketoconazole coadministration on electrocardiographic parameters in healthy volunteers.” J Clin Pharmacol. 37(11): 1065-72) have demonstrated that ketoconazole does not increase the QTc interval. This interval is used as a surrogate marker to determine whether drugs have the potential for inducing arrhythmia. US Patent Number 6,040,307 also makes reference to diminished hepatoxicity associated with the 2S,4R enantiomer but provides no data in support of that assertion. The method provided in US Patent Number 6,040,307 does not allow for the assessment of hepatoxicity as the method uses microsomes isolated from frozen tissue.
DIO-902 is the single enantiomer 2S,4R ketoconazole and is derived from racemic ketoconazole. It is formulated using cellulose, lactose, cornstarch, colloidal silicon dioxide and magnesium stearate as an immediate release 200 mg strength tablet. The chemical name is 2S,4R cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl] methoxyl]phenyl] piperazine, the formula is C26H28Cl2N4O4, and the molecular weight is 531.44. The CAS number is 65277-42-1, and the structural formula is provided below. The chiral centers are at the carbon atoms 2 and 4 as marked.
[0132]
Ketoconazole is an imidazole-containing fungistatic compound. DIO-902 is an immediate release tablet to be taken orally and formulated as shown in the table below.
Component
Percentage
2S,4R ketoconazole;
DIO-902
50%
Silicified Microcrystalline Cellulose, NF
(Prosolv HD 90)
16.5
Lactose Monohydrate, NF (316 Fast-Flo)
22.4
Corn Starch, NF (STA-Rx)
10
Colloidal Silicon Dioxide, NF (Cab-O-Sil M5P)
0.5
Magnesium Stearate, NF
0.6
The drug product may be stored at room temperature and is anticipated to be stable for at least 2 years at 25° C and 50% RH. The drug is packaged in blister packs.
A new process has been developed to separate ketoconazole (KTZ) enantiomers by membrane extraction, with the oppositely preferential recognition of hydrophobic and hydrophilic chiral selectors in organic and aqueous phases, respectively. This system is established by adding hydrophobic l-isopentyl tartrate (l-IPT) in organic strip phase (shell side) and hydrophilic sulfobutylether-β-cyclodextrin (SBE-β-CD) in aqueous feed phase (lumen side), which preferentially recognizes (+)-2R,4S-ketoconazole and (−)-2S,4R-ketoconazole, respectively. The studies performed involve two enantioselective extractions in a biphasic system, where KTZ enantiomers form four complexes with SBE-β-CD in aqueous phase and l-IPT in organic phase, respectively. The membrane is permeable to the KTZ enantiomers but non-permeable to the chiral selector molecules. Fractional chiral extraction theory, mass transfer performance of hollow fiber membrane, enantioselectivity and some experimental conditions are investigated to optimize the separation system. Mathematical model of I/II = 0.893e0.039NTU for racemic KTZ separation by hollow fiber extraction, is established. The optical purity for KTZ enantiomers is up to 90% when 9 hollow fiber membrane modules of 30 cm in length in series are used.
I, (−)-2S,4R-ketoconazole;
II, (+)-2R,4S-ketoconazole;
CDs, cyclodextrin derivatives;
l-IPT, l-isopentyl tartrate;
d-IPT, d-isopentyl tartrate;
HP-β-CD, hydroxypropyl-β-cyclodextrin;
Me-β-CD, methyl-β-cyclodextrin;
β-CD, β-cyclodextrin;
NTU, number of transfer units;
HTU, height of a transfer unit;
PVDF,polyvinylidene fluoride
…………………….
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-
Pelayo Camps, Xavier Farrés, Ma Luisa García, Joan Ginesta, Jaume Pascual, David Mauleón, Germano Carganico
Bromobenzoates (2R,4R)- and (2S,4S)-18, prepared stereoselectively from (R)- and (S)-epichlorohydrin, were transformed into (2R,4S)-(+)- and (2S,4R)-(−)-Ketoconazole, respectively, following the known synthetic protocols for the racemic mixture.
Tetrahedron Asymmetry 1995, 6(6): 1283
Stereoselective syntheses of both enantiomers of ketoconazole (1) from commercially available (R)- or (S)-epichlorohydrin has been developed. The key-step of these syntheses involves the selective substitution of the methylene chlorine atom by benzoate on a mixture of and or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
The incidence of fungal infections has considerably increased over the last decades. Notwithstanding the utility of the antifungal compounds commercialized in the last 15 years, the investigation in this field is however very extensive. During this time, compounds belonging to the azole class have beer, commercialized for both the topical and oral administrations, such a class including imidazoles as well as 1,2,4-triazoles. Some of these compounds car. show m some degree a low gastrointestinal tolerance as well as hepatotoxycity.
A large number of pharmaceutically active compounds are commercialized as stereoisomeric mixtures. On the other hand, the case in which only one of said stereoisomers is pharmaceutically active is frequent.
The undesired enantiomer has a lower activity and it sometimes may cause undesired side-effects.
Ketoconazole (1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine), terconazole (1-[4-[[2(2,4-dichlorophenyl)-2-[(1H-1 , 2 ,4-triazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine) and other related azole antifungal drugs contain in their structure a substituted 1,3-dioxolane ring, in which carbon atoms C2 and C4 are stereogenic centres, therefore four possible stereoisomers are possible. These compounds are commercialized in the form or cis racemates which show a higher antifungal activity than the corresponding trans racemates.
The cis homochiral compounds of the present invention, which are intermediates for the preparation of enantiomerically pure antifungal drugs, have been prepared previously in the racemic form and transformed into the different azole antifungal drugs in the racemic form [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979). J . Med . Chem . , 26, 611 (1983), J . Med . Chem . , 27 , 894 (1984) and US 4,144,346, 4,223,036, 4,358,449 and 4,335,125].
Scheme 1 shows the synthesis described for racemic ketoconazole [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979)]. Scheme 1
)
The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.
The preparation of racemic itraconazole [J. Heeres et al., J. Med . Chem. , 27 , 894 (1984)] is similar to that of terconazole, differing only in the nature of the phenol used in the last step of the synthetic sequence.
In the class of azoles containing a 1,3-dioxolane ring and a piperazine ring and moreover they are pure enantiomers, only the preparation of (+)- and (-)-ketoconazole has been described [D. M. Rotstein et al., J. Med . Chem . , 35, 2818 (1992)] (Scheme 2) starting from the tosylate of (+)- and (-) 2,2-dimethyl-1,3-dioxolane-4-methanol.
Scheme 2
This synthesis suffers from a series of drawbacks, namely: a) the use of expensive, high molecular weight starting products which are available only on a laboratory scale, and b) the need for several chromatographies during the process in order to obtain products of suitable purity, which maKes said synthesis economically unattractive and difficult to apply industrially.
Recently (N. M. Gray, WO 94/14447 and WO 94/14446) the use of (-)-ketoconazole and (+)-ketoconazole as antifungal drugs causing less side-effects than (±)-ketoconazole has been claimed.
The industrial preparation of enantiomerically pure antifungal drugs with a high antifungal activity and less side-effects is however a problem in therapy. The present invention provides novel homochiral compounds which are intermediates for the industrial preparation of already known, enantiomerically pure antifungal drugs such as ketoconazole enantiomers, or of others which have not yet been reported in literature, which are described first in the present invention, such as (+)-terconazole and (-)-terconazoie, which show the cited antifungal action, allowing to attain the same therapeutical effectiveness using lower dosages than those required for racemic terconazole
Example 14 : (2S,4R)-(-)-1-acetyl-4-[4-[ [2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine, (2S,4R) -(- )-ketoconazole.
This compound is prepared following the process described above for (2R,4S)-(+)-ketoconazole. Starting from HNa (60-65% dispersion in paraffin, 32 mg, 0.80 mmol), 1-acetyl-4-(4-hydroxyphenyl)piperazine (153 mg, 0.69 mol) and (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (250 mg, 0.61 mmol), upon crystallization from an acetone:ethyl acetate mixture, (2S,4R) -(-)-ketoconazole is obtained [(2S,4R)-V Ar = 2,4-dichlorophenyl, Y = CH, Z = COCH3] (196 mg, 61% yield) as a solid, m.p. 153-155ºC (lit. 155-157ºC); [α]D20 = -10.50 (c = 0.4, CHCl3) (lit. [α]D25 = -10.58. c = 0.4, CHCl3) with e.e. > 99% (determined by HPLC using the chiral stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1 % diethylamine as the eluent).
+ KETOCONAZOLE…. UNDESIRED
Example 7: (2 R ,4S)-(+)-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine (22, 4 S)-(+)-ketoconazole.
To a suspension of NaH (dispersed in 60-65% paraffin, 19.2 mg, 0.48 mmol) in anhydrous DMSO (3 ml),
1-acetyl-4-(hydroxyphenyl)piperazine (102 mg, 0.46 mmol) is added and the mixture is stirred for 1 hour at room temperature. Then, a solution of (2R,4R) – (+)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (160 mg, 0.39 mmol) in anhydrous DMSO (5 ml) is added, and the mixture is heated at 80ºC for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water
(20 ml) and extracted with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with water (3 × 25), dried with Na2SO4 and the solvent is evaporated off under vacuum. The oily residue thus obtained is crystallized from an acetone:ethyl acetate mixture to give (2R,4S)-(+)-ketoconazole ( (2R, 4 S) -V , Ar 2,4-dichlorophenyl, Y = CH , Z = COCH3 ) ( 110 mg , 5 3 % yie ld ) as a white solid, m.p. 155-156°C (lit. 154-156ºC), [α]D20 = + 8.99 (c = 0.4, CHCl3) (lit. [α]D25 = + 8.22, c = 0.4, CHCl3), with e.e. > 99% (determined by HPLC using the chirai stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1% of diethylamine, as the eluent; (+)-Ketoconazole retention time 73,28 min. (-)-Ketoconazole, retention time 79.06 min).
Experimental and theoretical analysis of the interaction of (+/-)-cis-ketoconazole with beta-cyclodextrin in the presence of (+)-L-tartaric acid
J Pharm Sci 1999, 88(6): 599
1H NMR spectroscopy was used for determining the optical purity of cis-ketoconazole enantiomers obtained by fractional crystallization. The chiral analysis was carried out using β-cyclodextrin in the presence of (+)-l-tartaric acid. The mechanism of the chiral discrimination process, the stability of the complexes formed, and their structure in aqueous solution were also investigated by 1H and 13C chemical shift analysis, two-dimensional NOE experiments, relaxation time measurements, and mass spectrometry experiments. Theoretical models of the three-component interaction were built up on the basis of the available NMR data, by performing a conformational analysis on the relevant fragments on ketoconazole and docking studies on the components of the complex. The model derived from a folded conformation of ketoconazole turned out to be fully consistent with the molecular assembly found in aqueous solution, as inferred from NOE experiments. An explanation of the different association constants for the complexes of the two enantiomers is also provided on the basis of the interaction energies.
Pantoprazole is a proton pump inhibitor drug used for short-term treatment of erosion and ulceration of the esophagus caused by gastroesophageal reflux disease.
Use
Pantoprazole is used for short-term treatment of erosion and ulceration of the oesophagus caused by gastroesophageal reflux disease. Initial treatment is generally of eight weeks’ duration, after which another eight week course of treatment may be considered if necessary. It can be used as a maintenance therapy for long term use after initial response is obtained.
Adverse effects
Antacid preparations such as pantoprazole work by suppressing the acid-mediated breakdown of proteins. This leads to an elevated risk of developing food and drug allergies due to undigested proteins passing into the gastrointestinal tract where sensitisation occurs. It is unclear whether this risk occurs with short-term or only long-term use.[1]
Nutrition: May reduce the absorption of important nutrients, vitamins and minerals, as well as medications, leaving users at increased risk for pneumonia.[2]
Cardiovascular: Increase in a chemical that suppresses the production of nitric oxide by 25% in humans, which have proven to relax and protect arteries and veins. Causes blood vessels to constrict, a development that could lead to a number of cardiovascular problems if continued for a prolonged period of time.[2]
Pharmacology
Wyeth pantoprazole 20mg.
Pantoprazole is metabolized in the liver by the cytochrome P450 system.[3] Metabolism mainly consists of demethylation by CYP2C19followed by sulfation. Another metabolic pathway is oxidation by CYP3A4. Pantoprazole metabolites are not thought to have any pharmacological significance. Pantoprazole is relatively free of drug interactions;[4] however, it may alter the absorption of other medications that depend on the amount of acid in the stomach, such as ketoconazole or digoxin. Generally inactive at acidic pH of stomach, thus it is usually given with a pro kinetic drug. Pantoprazole binds irreversibly to H+K+ATPase (proton pumps) and suppresses the secretion of acid. As it binds irreversibly to the pumps, new pumps have to be made before acid production can be resumed. The drug’s plasma half-life is about 2 hours.[5]
Pharmacokinetics
Absorption
Bioavailability: (oral, delayed release tablets), approximately 77%
Effect of food: (oral, delayed-release tablets), AUC and Cmax no effect, Tmax variable, absorption delayed, no net effect
Effect of food: (oral, for-delayed-release suspension), administer 30 minutes before a meal
Tmax, Oral, delayed-release suspension: 2 to 2.5 h
Tmax, Oral, delayed-release tablets: 2.5 h
Tmax, Oral, delayed-release tablets: 1.5 to 2 hours (pediatrics)
Distribution
Protein binding: about 98% to primarily albumin
Vd, extensive metabolizers (IV): approximately 11 L to 23.6 L
Vd, pediatrics (oral): 0.21 to 0.43 L/kg.
Metabolism
Hepatic; cytochrome P450 CYP2C19; minor metabolism from CYP3A4, 2D6, and 2C9
Excretion
Fecal: (oral or IV, normal metabolizers), 18%
Renal: (oral or IV, normal metabolizers), approximately 71%, none as unchanged
Dialyzable: no (hemodialysis)
Total body clearance: (IV) 7.6 to 14 L/hour.
Total body clearance: (oral, pediatrics) 0.18 to 2.08 L/h/kg
Elimination Half Life
Oral or IV, 1 hour
Oral or IV, slow metabolizers, 3.5 to 10 hours
Pediatrics, 0.7 to 5.34 hours
Availability
Pantoprazole was developed by Altana (owned by Nycomed) and was licensed in the USA to Wyeth (which was taken over by Pfizer). It was initially marketed under the brand name Protonix by Wyeth-Ayerst Laboratories and now is available as a generic. It is available by prescription in delayed-release tablets. It is also available for intravenous use.
On 24 December 2007, Teva Pharmaceutical released an AB-rated generic alternative to Protonix.[6] This was followed by generic equivalents from Sun Pharma and Kudco Pharma. Wyeth sued all three for patent infringement and launched its own generic version of Protonix with Nycomed.[7][8]
Pantoprazole is the international non-proprietary name of the chemical product 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2- pyridinyl)methyl]sulfmyl]-lH-benzimidazole of formula
Pantoprazole This product is an active ingredient used in the treatment of gastric ulcers, usually in the form of its sodium salt.
The product was described for the first time in European patent application EP-A-0166287 that also describes several processes for the preparation of products assignable to a general formula among which pantoprazole is to be found. The reaction sequences of these processes, applied precisely to the preparation of pantoprazole, are given in Scheme 1.
Scheme 1
In Scheme 1, the variables Y, Z, Z’ and Z” are leaving groups, for example atoms of halogen, and the variables M and M’ are atoms of alkali metals.
Austrian patent AT-B-394368 discloses another process based on a different route of synthetis, the reaction sequence of which is given in Scheme 2.
Pantoprazole Scheme 2
Nevertheless, this process has obvious drawbacks, since the methylation can take place not only in OH in the 4-position of the pyridine ring, but also in the nitrogen linked to a hydrogen of the benzimidazole ring, which can give place to mixtures of the desired product with the two possible methylated isomers of the benzimidazole compounds obtained, 3- methyl or 1 -methyl, which means that additional chromatographic purification steps are needed and the yields obtained are low.
PCT application WO97/29103 discloses another process for the preparation of pantoprazole, the reaction sequence of which is given in Scheme 3.
Scheme 3 As may be seen, different synthesis strategies have been proposed for the preparation of pantoprazole, some of them recently, which is an indication that the preparation of the product is still not considered to be sufficiently well developed, whereby there is still a need for developing alternative processes that allow pantoprazole to be prepared by means of simpler techniques and more accessible intermediate compounds and with good chemical yields.
EXAMPLES
Example 1. – Preparation of compound (IX)
47.5 ml (0.502 mol) of acetic anhydride were mixed with 1.65 g (0.0135 mol) of 4-dimethylaminopyridine, giving a transparent yellow solution which was heated to 65° – 70°C. This temperature was held by cooling since the reaction is exothermic. 25 g (0.1441 mol) of 2-methyl-3- methoxy-4-chloropyridine N-oxide (X) were added over a period of about 70 minutes. Once the addition was completed, the reaction was held at 65° – 70°C for a further 2h 20 minutes and after this time it was allowed to cool down to below 65°C and 90 ml of methanol were added gradually, while holding the temperature below 65°C. The resulting reaction mass was distilled at reduced pressure in a rotavap to remove the volatile components and the residue containing compound (IX) was used as such for the following reaction. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), showed a single spot at Rf – 0.82, indicating that the reaction has been completed.
Example 2. – Preparation of compound fVIII
(IX) (VIII)
11.5 ml methanol and 11.5 ml of water were added over the crude residue from Example 1 containing compound (IX), and thereafter, while holding the temperature to between 25° and 30°C with a water bath, the residual acetic acid contained in the crude residue was neutralized by the addition of 33% aqueous NaOH. Once the residual acid had been neutralized, 19 ml (0.2136 mol) of the 33% aqueous NaOH were added over 20 minutes, while holding the temperature to between 25° and 30°C, and, on completion of the addition, the hydrolysis reaction at pH 11.7 – 11.8 was held for 2h 30 minutes, to between 25° and 30°C. On completion of the reaction, the pH was adjusted to 7.0 – 7.5 by the addition of HC1 35%, while holding the temperature to 25°C. Thereafter, 50 ml of methylene chloride were added and, after stirring and allowing to rest, the phases were decanted. A further five extractions were carried out with 30 ml methylene chloride each and the pooled organic phases were dried with anhydrous sodium sulfate, were filtered and washed, and were evaporated at reduced pressure in a rotavap, providing a solid residue having a melting point around 73°C and containing compound (VIII). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.55, showing that the reaction was complete. The thus obtained crude residue was used as such in the following reaction.
Example 3. – Preparation of compound (VI)
24.5 g of the residue obtained in Example 2, containing approximately 0.142 mol of the compound 2-hydroxymethyl-3-methoxy-4-chloropyridine (VIII), were mixed with 0.5 ml of DMF and 300 ml of anhydrous methylene chloride, to give a brown solution which was cooled to 0° – 5°C in an ice water bath. Thereafter, a solution of 11.5 ml (0.1585 mol) of thionyl chloride in 50 ml of anhydrous methylene chloride was added over 20 minutes, while holding the above-mentioned temperature,. Once the addition was complete, the reaction was held at 0° – 5°C for a further 90 minutes and then 120 ml of water and NaOH 33% were added to pH 5 – 6, requiring approximately 29 ml of NaOH. The phases were then decanted and separated. The organic phase was extracted with a further 120 ml of water and the pooled aqueous phases were extracted with a further 4×25 ml of methylene chloride, in order to recover the greatest possible amount of product. The pooled organic phases were dried over anhydrous sodium sulfate, filtered and washed, and evaporated at reduced pressure in a rotavap, to give a residue containing the compound 2-chloromethyl-3- methoxy-4-chloropyridine (VI). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15:1), showed a main spot at Rf = 0.83, indicating that the reaction was complete. The thus obtained crude residue was used as such in the following reaction. Example 4. – Preparation of compound (III)
26.11 g of the residue obtained in the Example 3 containing approximately 0.136 mol of the compound 2-chloromethyl-3-methoxy-4- chloropyridine (VI) were mixed with 370 ml of methylene chloride, to give a brown solution over which were added, at 20° – 25°C, 29.3 g (0.136 mol) of 5-difluoromethoxy-2-mercaptobenzimidazole (VII) and 17.10 ml (0.136 mol) of tetramethylguanidine (TMGH). The mixture was stirred at this temperature for 2 hours, after which 450 ml of water were added, with the pH being held to between 9.5 and 10. Thereafter the phases were decanted and the organic phase was washed 5×50 ml of a IN NaOH aqueous solution and, thereafter, with 2×50 ml of water. The organic phase was treated with 50 ml of water and an amount of HC1 30% sufficient to adjust the pH to between 5 and 6. Thereafter, the phases were decanted, and the organic phase was dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a solid residue of melting point 64° – 73 °C that contains the compound (III). Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), presented a main spot at Rf = 0.52. Yield 82%. The thus obtained compound 5-(difluoromethoxy)-2-[[(3-methoxy-4-chlorine-2 pyridinyl)methyl]mercapto]- lH-benzimidazole (III) was used as such in the following reaction Example 5. – Preparation of compound (IV)
25.8 g (0.0694 mol) of the compound (III) obtained in the Example 4 were mixed with 88 ml of methanol, to give a brown solution to which 3.7 ml of water, 0.99 g of ammonium molybdate and 0.78 g of sodium carbonate were added. The system was cooled to 0°C – 5°C, 3.4 ml (0.0756 mol) of 60% hydrogen peroxide were added, and the reaction mixture was held at 0°C – 5°C for 1 – 2 days, the end point of the reaction being checked by thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: l).
During the reaction the presence of hydrogen peroxide in the reaction medium was controlled by testing with potassium iodide, water and starch. When effected on a sample containing hydrogen peroxide, it provides a brown-black colour. If the assay is negative before the chromatographic control indicates completion of the reaction, more hydrogen peroxide is added.
On completion of the reaction, 260 ml of water were added, the system was cooled to 0°C – 5°C again and the mixture was stirred for 2 hours at this temperature. The solid precipitate was filtered, washed with abundant water, and dried at a temperature below 60°C, to give 5-(difluoromethoxy)-2-[[(3- methoxy-4-chlorine-2-pyridinyl)methyl]sulfinyl]-lH-benzimidazole (IV), melting point 130° – 136°C, with an 83.5% yield. Thin layer chromatography on silica gel 60 F254, eluting with CHCl3/MeOH (15: 1), gave a main spot at Rf = 0.5.
Compound (IV) can be purified, if desired, by the following crystallization method:
5 g of crude product was suspended in 16 ml of acetone and was heated to boiling until a dark brown solution was obtained. Thereafter the thus obtained solution was allowed to cool down to room temperature and then was then chilled again to -20°C, at which temperature the mixture was held for 23 hours without stirring. Thereafter the solid was filtered and washed with 6×4 ml of acetone chilled to -20°C. Once dry, the resulting white solid weighed 2.73 g, had a point of melting of 142°C and gave a single spot in thin layer chromatography. The IR spectrum of the compound on KBr is given in Figure 1.
The acetonic solution comprising the mother liquors of filtration and the washes was concentrated to a volume of 20 ml and a further 5 g of crude compound were added. The above described crystallization process was repeated to obtain a further 4.11 g of purified product of characteristics similar to the previous one.
The acetonic solution from the previous crystallization was concentrated to a volume of 17 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 2.91 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 15 ml and a further 4 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.3 g of purified product of similar characteristics to the previous ones.
The acetonic solution from the previous crystallization was concentrated to a volume of 16 ml and a further 4.36 g of crude compound were added. The above described crystallization process was repeated to obtain a further 3.62 g of purified product of similar characteristics to the previous ones.
Finally, the acetonic solution from the previous crystallization was concentrated to a volume of 10 – 12 ml and held at -20°C for two days without stirring. Thereafter, the solid was filtered and washed with 5×3 ml of acetone chilled to -20°C. Once dry, the solid weighed 1.26 g and had similar characteristics to the previous ones.
The total yield of all the crystallizations was 80%.
Example 6. – Preparation of pantoprazole
12.95 g (0.0334 mol) of compound (IV) purified by crystallization of Example 5 were mixed with 38 ml of N,N-dimethylacetamide and thereafter 7.03 g (0.1003 mol) of potassium methoxide were added, while holding the temperature to between 20°C and 30°C, whereby a dark brown mixture was obtained. The system was held at approximately 25°C for about 23 hours, after which, once the reaction was complete, the pH was adjusted to 7 with the addition of 3.82 ml of acetic acid. The N,N-dimethylacetamide was removed at reduced pressure at an internal temperature of not more than 75°C. 65 ml of water and 50 ml of methylene chloride were added over the thus obtained residue, followed by decantation of the phases. Once the phases were decanted, the aqueous phase was extracted a with further 3×25 ml of methylene chloride, the organic phases were pooled and the resulting solution dried over anhydrous sodium sulfate, was filtered and washed, and evaporated at reduced pressure in a rotavap, to give a crude residue over which 55 ml of water were added, to give a suspension (if the product does not solidify at this point the water is decanted and a further 55 ml of water are added to remove remains of N,N-dimethylacetamide that hinder the solidification of the product). The solid was filtered and, after drying, 11.61 g of crude pantoprazole of reddish brown colour were obtained (Yield 90%). The thus obtained crude product was decoloured by dissolving the crude product in 150 ml of methanol, whereby a dark brown solution was obtained. 7.5 g of active carbon were added, while maintaining stirring for 45 minutes at 25°C – 30°C, after which the carbon was filtered out and the filter was washed. The methanol was then removed in the rotavap at reduced pressure, a temperature below 40°C. 10.33 g of a solid residue were obtained and were mixed with 14.9 ml of methylethylketone, and the suspension was heated to 45°C for about 10 minutes, after which it was cooled, first to room temperature and then to -20°C. This temperature was held over night and thereafter the solid was filtered, washed with 6×5 ml of methylethylketone chilled to -20°C. Once dry, 7.75 g of a white solid, melting point 140°C – 141 °C, were obtained. Thin layer chromatography on silica gel F254, eluting with CHCl3/MeOH (15: 1), gave a single spot at Rf =
0.41 and a IR spectrum corresponding identically with that of pantoprazole.
The ketonic solution comprising the mother liquors of filtration and the washes, was concentrated to 9.7 ml, was heated to 40°C, was held at this temperature for about five minutes and was then cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 4×2 ml of methylethylketone chilled to -20°C. Once dry, 0.42 g of a white solid of similar characteristics to the previous one was obtained.
The ketone solution from the previous treatment was concentrated to 3.1 ml, was heated to 40°C, was held to this temperature for about five minutes and then was cooled, first to room temperature and then to -20°C, this temperature being held for 4 hours. At the end of this time, the solid was filtered and was washed with 5×3 ml of methylethylketone chilled to – 20°C. Once dry, 0.41 g of a white-beige solid of similar characteristics to the previous one was obtained. The total yield, including purifications, was 67%.
If a whiter solid is desired, one or several washes can be carried with isopropyl acetate as follows: 6.6 g of pantoprazole from the methylethylketone treatment were suspended in 50 ml of isopropyl acetate. The system (white suspension) was stirred for about 30 minutes at 25°C, was then cooled to 0°C – 5°C, was stirred for about 15 minutes at this temperature and the solid was then filtered, was washed with 3×15 ml of isopropyl acetate. Once dry, 6.26 g of a pure white solid were obtained.
Trade Names
Country
Trade name
Manufacturer
Germany
Pantozol
Nycomed
Rifun
- “-
France
Eupantol
Altana
Inipomp
Sanofi-Aventis
United Kingdom
Protium
ALTANA
Italy
Pantekta
Abbott
Pantopan
Pharmacia
Pantork
Altana
USA
Protonix
Wyeth
Ukraine
Kontrolok
Nycomed Oranienburg GmbH, Germany
Nolpaza
Krka
Pultset
Nobel Ilach Sanayi ve Ticaret AS, Turkey
Proksium
JSC “Lubnyfarm”, Ukraine
various generic drugs
Formulations
ampoule 40 mg;
Tablets 40 mg
UV – spectrum
Conditions : Concentration – 1 mg / 100 ml
Solvent designation schedule
Methanol
Water
0.1 M HCl
0.1M NaOH
The absorption maximum
289 nm
291nm
Observed
decay
295 nm
391
346
-
418
ε
16600
14700
-
17700
IR – spectrum
Wavelength (μm)
Wavenumber (cm -1 )
NMR Spectrum
will be added
Links
EP 134 400 (Byk Gulden Lomberg; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,555,518 (Byk Gulden Lomberg; 26.11.1985; appl. 1.5.1984; CH-prior. 3.5.1983).
US 4,758,579 (Byk Gulden Lomberg; 19.7.1988; appl. 28.4.1987; CH-prior. 16.6.1984).
UV and IR Spectra. H.-W. Dibbern, RM Muller, E. Wirbitzki, 2002 ECV
NIST / EPA / NIH Mass Spectral Library 2008
Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman.Academic Press, 2000.
Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
[Dr. John Cooke, chair of Methodist Hospital's cardiovascular services] [Houston Chronicle Health Zone dated Thursday, July 11, 2013 chron.com/refluxmeds] (Journal: Circulation)
Jump up^ Meyer, U A (1996). “Metabolic interactions of the proton-pump inhibitors lansoprazole, omeprazole and pantoprazole with other drugs”. European journal of gastroenterology & hepatology8 (Suppl 1): S21–25. doi:10.1097/00042737-199610001-00005.
Steinijans, V. W.; Huber, R.; Hartmann, M.; Zech, K.; Bliesath, H.; Wurst, W.; Radtke, H. W. (1996). “Lack of pantoprazole drug interactions in man: An updated review”. International Journal of Clinical Pharmacology and Therapeutics34 (6): 243–262. PMID8793611.
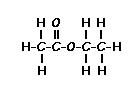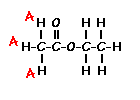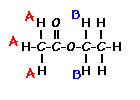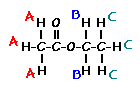
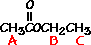


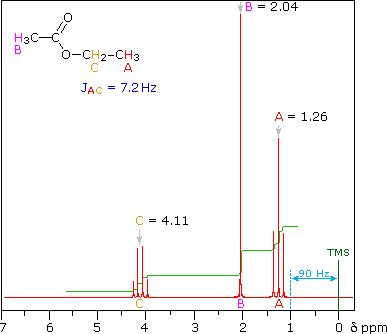
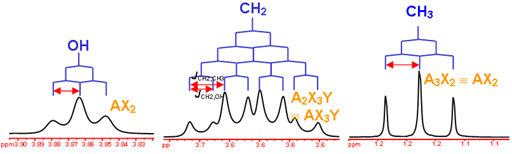



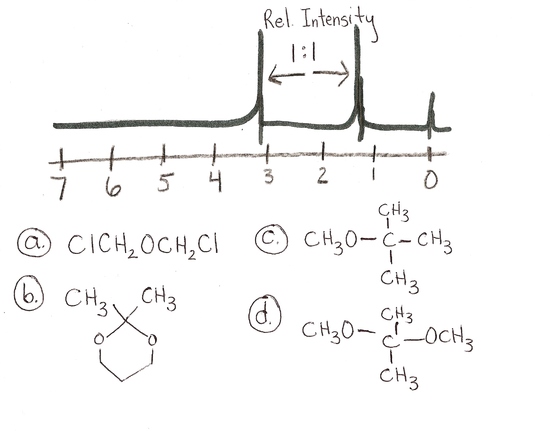 answer is a and d
answer is a and d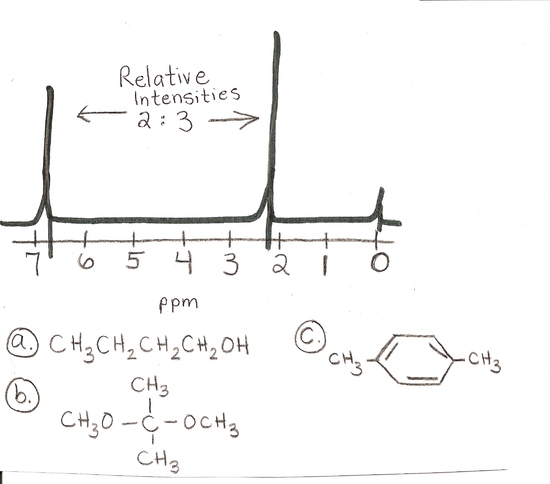
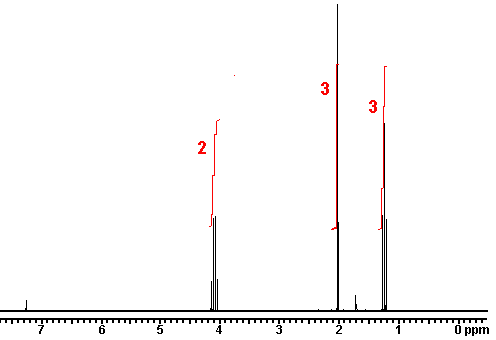
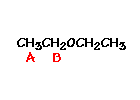
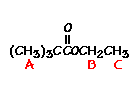






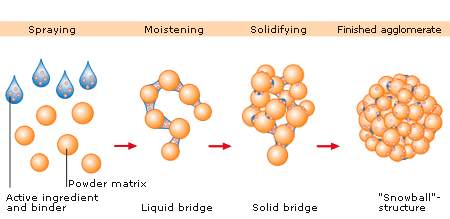






 Dedicated to all moms
Dedicated to all moms

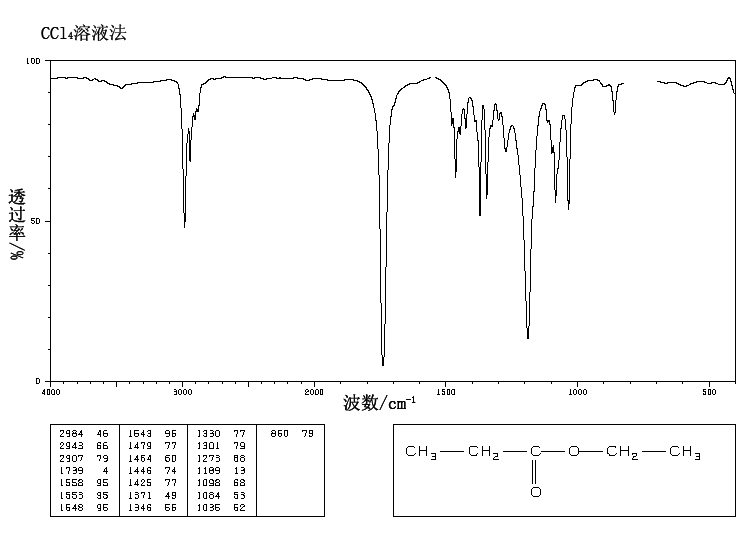
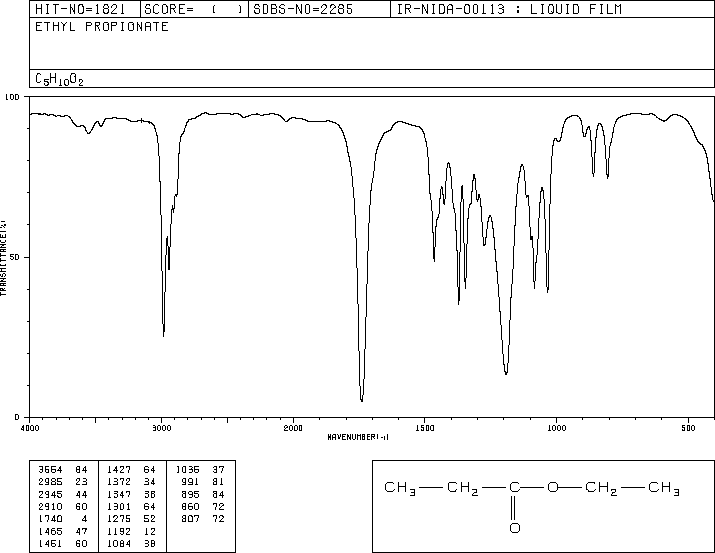


 This is the structure. See if you can assign the peaks on your own.
This is the structure. See if you can assign the peaks on your own. C has a higher chemical shift than D because it’s closer to a more electron-withdrawing functional group.
C has a higher chemical shift than D because it’s closer to a more electron-withdrawing functional group.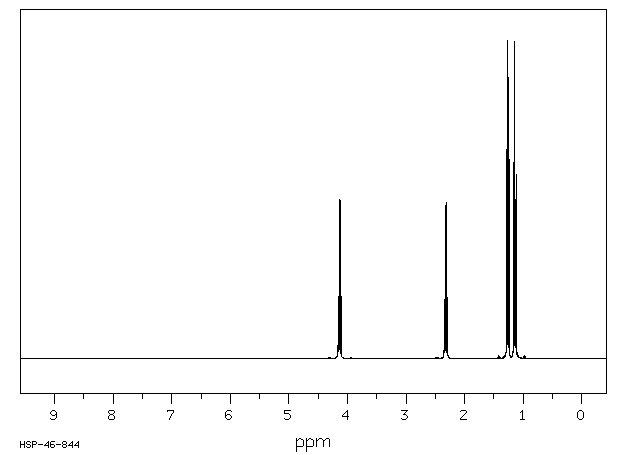

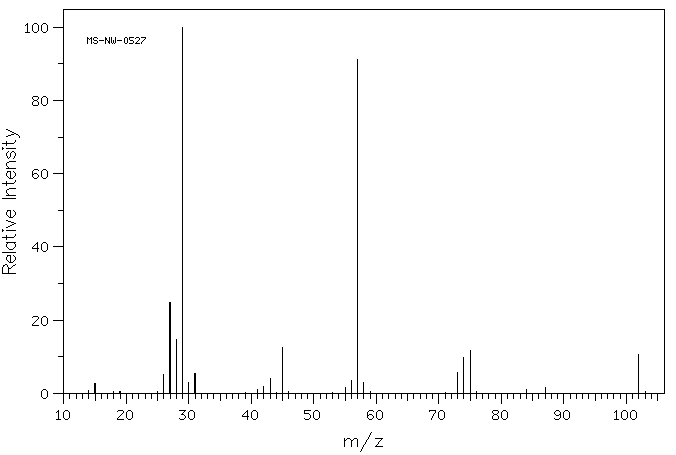
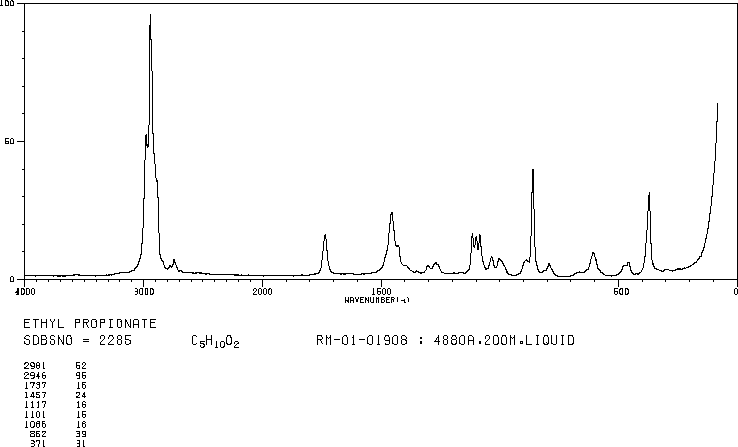


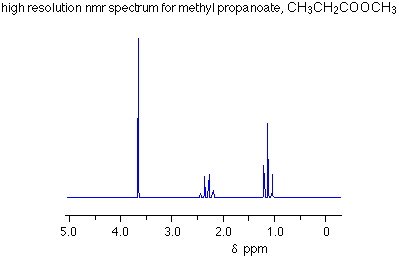



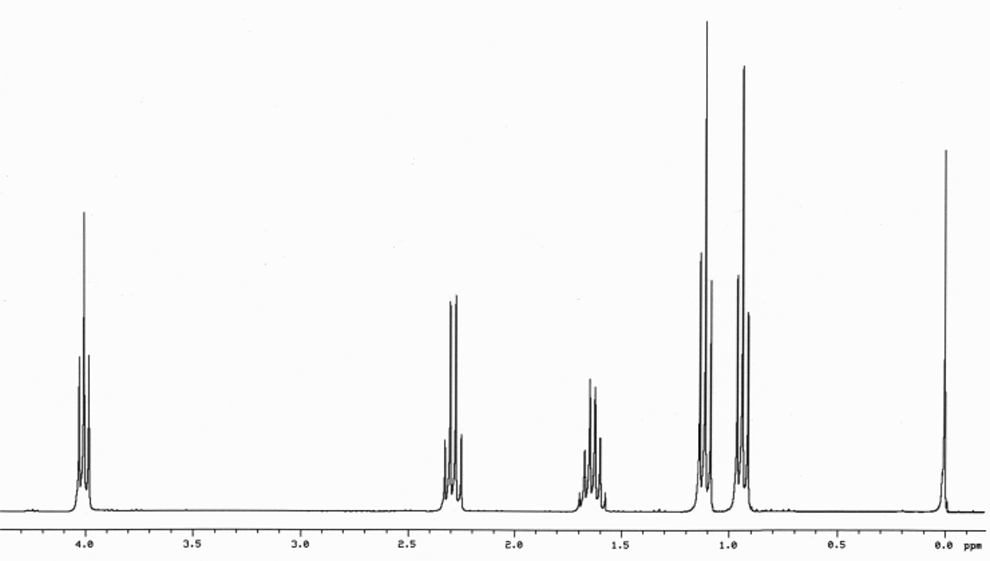









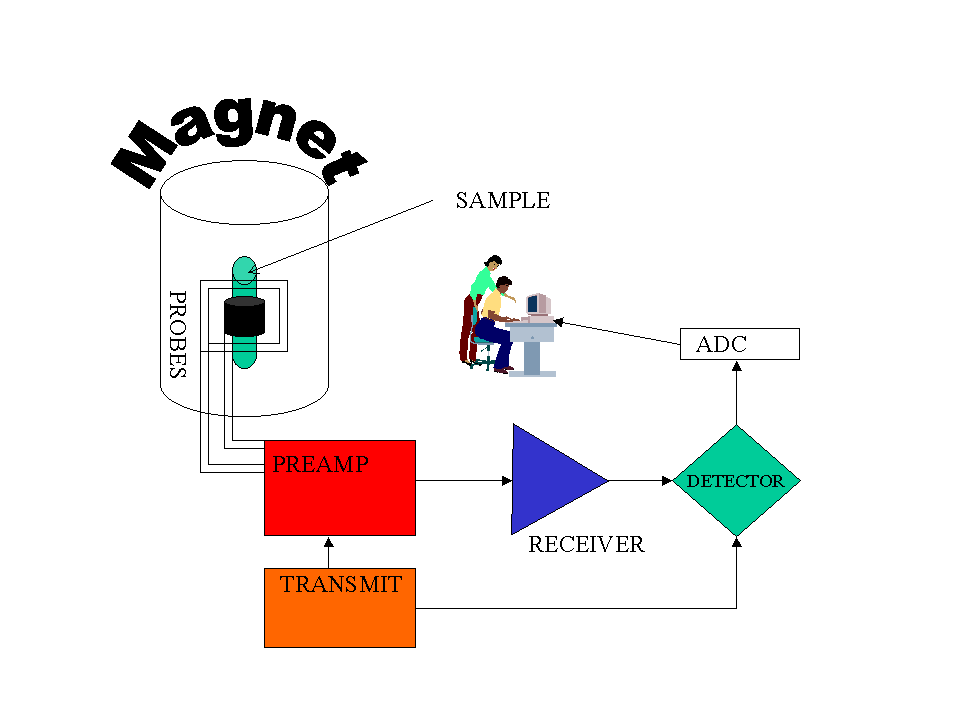







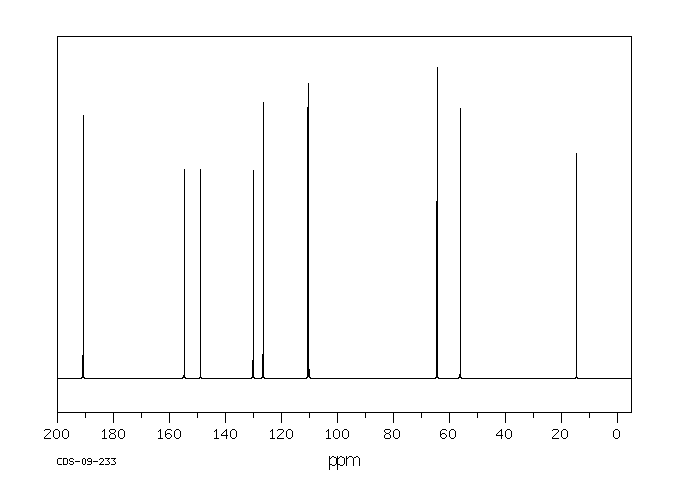
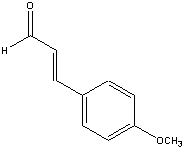
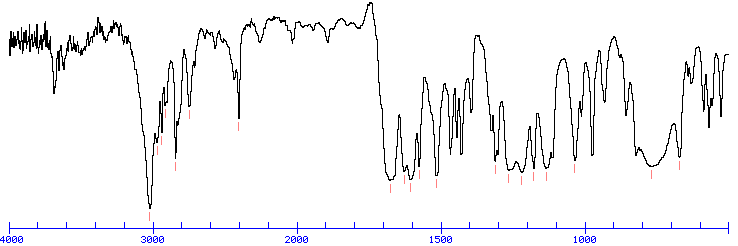
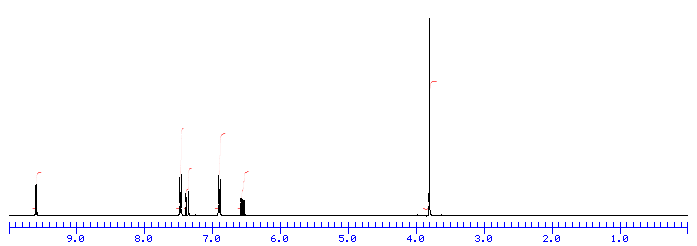


















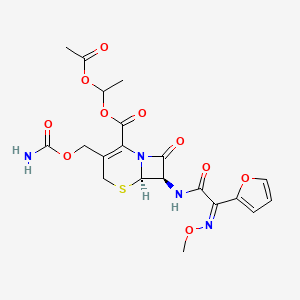





























 and
and  or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.