![]()
Vinita Gupta, 43, Group President and CEO, Lupin Pharmaceuticals and Director, Lupin
Feb 2015….India-based drugmaker Lupin has signed an agreement with Polish biopharmaceutical firm Celon Pharma to develop a fluticasone / salmeterol dry powder inhaler (DPI).
Under the deal, Lupin will take the responsibility for commercialisation of the product, which is a generic version of GlaxoSmithKline’s (GSK) Advair Diskus.
Lupin CEO Vinita Gupta said: “We are very pleased to partner with Celon given their experience in the development and manufacturing of fluticasone/salmeterol DPI in Europe…………..http://www.pharmaceutical-technology.com/news/newslupin-celon-pharma-partner-generic-version-gsks-advair-diskus-4514718?WT.mc_id=DN_News
Ms. Vinita Gupta is the CEO of Lupin Pharmaceuticals Inc, USA, (LPI) and Group President, Director on the board of Lupin Limited and a Director on the Board of Lupin’s Japanese subsidiary Kyowa Pharmaceuticals. Ms. Gupta is responsible for the North American and European business of the company.
Ms. Gupta joined Lupin in 1992 and developed Lupin’s entry strategy into US and Europe. Under her leadership Lupin has emerged as a leader in the US generic market as well as the only company from India to have a successful brand business in the US. As part of her responsibility she built the entire management team for the US and European business and supervised the development of the company’s pipeline.
Ms. Gupta holds a Bachelor’s degree in Pharmacy from the University of Mumbai and MBA from J L Kellogg Graduate School of Management, Northwestern University.
“A good year” is how Vinita Gupta, Group President and CEO at Lupin Pharmaceuticals, describes her company’s performance at a time when unsettling news was the key takeaway for pharma companies. Lupin grew by an impressive 35.9 per cent globally and 24 per cent in India. New product launches helped it grow its generics business by 52 per cent, making it the sixth-largest generics pharmaceutical company globally by market capitalisation and the third-largest Indian pharmaceutical company by revenues. “I can’t think of any challenges that affected Lupin’s performance during the last fiscal year,” says Gupta, 44. The company’s strategy now is to focus strongly on building its branded business globally.
![]()
Vinita Gupta, 43, Group President and CEO, Lupin Pharmaceuticals and Director, Lupin, is based in the United States, but has been in India a lot in the past one year.
With an expanding role in Lupin’s universe, Vinita has been spending more time outside the US, at times taking her six-year-old son, Krish with her. “He is getting exposure at a much younger age,” she says. Gupta herself was exposed to business at the age of 11 by her father Desh Bandhu Gupta, Lupin’s founder and Chairman.
“We almost had a family board at home, discussing work,” she says. Currently work goes well indeed, with Gupta taking new initiatives in India and also making the business more global. “I am focusing on drivers for growth in our business for the next five years,” she says.
Gupta is married to US-based businessman Brij Sharma.
She was 13 when she travelled to Switzerland with her father, to watch him position the family-run Lupin Limited, to negotiate and to strategise. It was enough to get her hooked. Enough for her to move away from a childhood fascination for art and enter the world of pharmaceuticals. Group president and CEO, Lupin Pharmaceuticals, and director on the board of Lupin Limited, Vinita Gupta has never regretted that decision. The 41-year-old is responsible for creating a substantial international presence for the company that was born in Mumbai in 1968 and named after a leguminous flower.
The Lupin group produces affordable generic and branded formulations in the world with a significant presence in cardiovasculars, diabetes, asthma, pediatrics and anti-infectives. But Desh Bandhu Gupta, her father, wanted the company to make an impact in the western market.
It was a challenge that seemed perfect for Gupta who graduated in pharmacy from the University of Mumbai and then spent a year working at the company in Mumbai. She then moved to the US for an MBA from the J.L. Kellogg Graduate School of Management at Northwestern University, following it with a brief stint in a US pharma company. But she didn’t want to be a mere “cog in the wheel”, returning to India to take up that initial challenge-to create a business strategy that would allow Lupin to enter American and European markets.
Today, Lupin is the ninth largest generics company in the US. It is also one of India’s top five pharmaceutical players and one of the fastest growing top 10 generic players in Japan and South Africa. The US arm of the business, Gupta’s baby, contributes to over 30 per cent of Lupin’s revenues, a company that clocked in close to Rs.4,000 crore in 2008-2009. Nine out of the 23 generic products Lupin has in the US market are at the number one position giving consistent competition to larger US pharma companies.

With brother NileshThe beginning however was difficult. After all, India wasn’t very well known in the US market. “We realised that we had the aspirations but not the infrastructure in the form of facilities to meet US and European requirements and standards,” she remembers. So she spent three years building the infrastructure, creating a process that would be acceptable to these regulated markets. The break came with Suprax, a pediatric antiinfective drug that was valued at nearly $60 million in the US market. Gupta had already filed for a generic of the brand. “Suddenly, we had the opportunity to brand the generic, so we licensed the brand name from the innovator as he had left the market,” she remembers. It was a three-person team with 40 outsourced sales people.
Today, the product’s sales are at $74 million. It has been satisfying, she says. “The innovator was in the market with a sales force of 300 people. We are 60.” The aim, she says, has always been to balance branded products as well as generic. The success, her father and chairman of the company, Desh Bandhu Gupta, says, stems not just from her determination. “It’s also her intimate understanding of the entire pharma spectrum with the motivation to see it through,” he says.
This determination became obvious when she managed to persuade the dean at Kellogg to give her admission, even though she was 19 and perhaps the youngest in her class. It was a challenging time as she learned to balance her work and household chores. “At that time in India, everything was handwritten. I had to do every single thing using the computer,” says Gupta who often bribed her friends with homemade Indian food to type out her projects. It taught her to be independent.
But it was perhaps, two months ago, when Gupta a bigger challenge. A deal that made tough seem an understatement. For Antara, an anti-cholesterol drug. “It was very much like Suprax, that was serendipity,” says Gupta. It was a large product with high potential. But the company was in bankruptcy. “I was sure we could do things differently with the product,” she says.
Gupta says Lupin was the first to file for the generic brand. But they couldn’t own the generic and the brand. She had six weeks to sell the generic, win the bid in the US bankruptcy court and buy the brand. She did it. At $38 million, one-third its market value. It’s a deal that Nilesh, her brother and group president and executive director, believes displayed his sister’s meticulous calibre. “There were three sets of negotiations going on at the same time. And while there were others involved, this deal was Vinita all the way,” he says.
Kamal Sharma, managing director, Lupin Limited, has watched Gupta transform from a teenager learning the ropes of a business to the successful go-getter that she is today. “She values, teaches and encourages her people to deliver consistent results year after year,” he says. It’s an attitude that is apparent from the get-go.
 With Richa, Kavita, Anuja and Nilesh")
(L-R) With Richa, Kavita, Anuja and NileshAt the Trident, Mumbai, for the photo shoot, Gupta is comfortable surrounded by people, even though she is a little hesitant in front of the camera. It’s here that she actually seems to shed the image of an ultimate powerhouse, a businesswoman driven to succeed. It is here that she becomes the Mumbaikar who prefers a masala chai over brewed coffee and a plain tee over a designer label. In some ways, she is still the girl who grew up in a housing society near the airport in Vile Parle, Mumbai. It’s the kind of place where people still keep their doors open and where one can walk into a neighbour’s house without having to knock. Things weren’t handed to her on a golden platter, she admits. In fact, she says, their father taught them that, “as a family we would have to work harder to earn and deserve our right more than what other professionals do.”
As a child, she remembers sharing a room with her four siblings, Kavita, Anuja, Nilesh and Richa. She didn’t like that very much. “But now, when I think of it, I feel it was an amazing life,” she says. Her father adds that he always took his children to different countries, either on work or otherwise. It was his way of showing them the world and different experiences.
But a different side emerges as Gupta talks of the pharma industry. “I dreamed of taking what Dad had built and adding value to it in the western markets,” she says. “This is what I had always prepared myself for. I am living the dream.” And it isn’t as if there aren’t any downs. Six months ago, she remembers, the company received a warning letter from the Food and Drug Administration. She spent that time working to resolve their concerns. “And then three months later, we made one of our most attractive acquisitions. The industry is so quick changing, so dynamic. It always keeps you thinking,” she says.
For Nilesh though, Gupta is his sounding board, the eldest sister with whom he shares a relationship that complements their work profiles. And while Nilesh says with a laugh, Gupta doesn’t pull the bigsister act with him at work, home is a different story. Gupta admits with a mock sigh, “You can’t posture with your siblings. You can posture with anyone else, but not your siblings.”

With husband Brij and son KrishThe obvious downside, however, is family. Her work keeps her busy, sees her up and in office by 8 am, back just in time to spend about an hour with her four-year-old son Krish. “He was a very easy child till some time ago, but lately he has become very demanding,” she says with a smile. Just as Gupta was leaving her home in Baltimore, Maryland for her current trip to India, Krish demanded they go leaf-picking in their backyard. “More than anything, I loved watching the expression on his face while we were picking leaves. His smile brightens up my day,” she says.
As much as her job is a passion she tries to spend time with Krish and husband Brij Sharma, a businessman whom she met in the US. “My husband is a very good listener. I keep talking whenever I am with him and he listens even today,” she says with a laugh. A workout is a must, however, as Gupta heads to the gym every day, spinning the cycle even when she was eight months pregnant.
“My husband jokes that’s the reason why Krish thinks and behaves ahead of his age,” laughs Gupta. But biking near the waterfront with her son and spending time on her husband’s boat is an activity that wins hands down. As does time spent with her two sisters Anuja and Richa, who live in Chicago. While Anuja is a pediatric cardiologist, Anuja is into public health. They do plan vacations together, but she often discovers that her brother Nilesh refuses to talk to her over the weekend. “Probably because I always end up talking about work,” she says with a laugh. “It has become so much a part of our lives,” she says.
![]()
Biggest Challenge
To bring in the changes required that will continue to set the company apart from the competition, and to attain a good work-life balance
In June2012 , Vinita Gupta, CEO of Lupin Pharmaceuticals Inc, the Indian drug maker’s US unit, received the “Entrepreneur of the Year” award from Ernst & Young in the health services and technology category for Maryland state of the US. Over the past year, the US business of Lupin crossed the $500 million mark.
ms-vinita-gupta-ceo-lupin-pharma-winner-of-the-e-y-entrepreneur-of-the-year-2012-award Ms Vinita Gupta, CEO, Lupin Pharma – Winner of the E&Y Entrepreneur of the Year 2012 Award Singapore: Ernst & Young LLP have awarded the Entrepreneur of the Year 2012 Award in the Health Services and Technology category to Ms Vinita Gupta, CEO, Lupin Pharmaceuticals. As a Maryland award winner, Ms Gupta is now eligible for consideration for the National Entrepreneur of the Year 2012 Award. The award recognizes outstanding entrepreneurs, who demonstrate excellence and extraordinary success in such areas as innovation, financial performance and personal commitment to their businesses and communities. Ms Gupta was selected by an independent panel of judges, and the award was presented at a special gala on June 28, 2012, at the Baltimore Marriott Waterfront, Maryland. Commenting on the award, Ms Vinita Gupta, CEO, Lupin Pharmaceuticals said, “I am honored to receive this recognition on behalf of our company. We have been very fortunate to have multiple opportunities to grow and differentiate our organization while bringing quality, affordable generics and valuable brands to the US market. The passion, dedication and entrepreneurial spirit of our team has set us apart from competition.”
![]()
DB Gupta (centre) Chairman, Vinita Gupta (right) CEO and Nilesh Gupta
![Share]()















 Charlotte
Charlotte 












 PIC CREDIT TO………..
PIC CREDIT TO……….. 


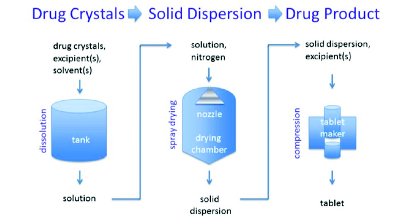



































 thanks to cornell
thanks to cornell Cornell law school
Cornell law school Cornell law school
Cornell law school












 ME
ME1612-1880/asset/cover.gif?v=1&s=cc68b12e733d21d6415eeec10551f83cb03996eb "Chemistry & Biodiversity")
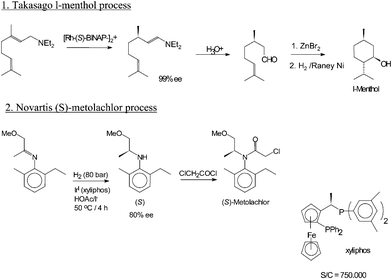









 Edmunds Lukevics
Edmunds Lukevics












")
")
")
")
")



 With Richa, Kavita, Anuja and Nilesh")



 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 amcrasto@gmail.com
amcrasto@gmail.com

 CHINA
CHINA


















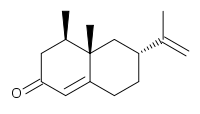


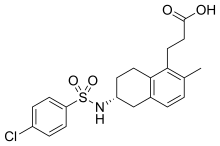
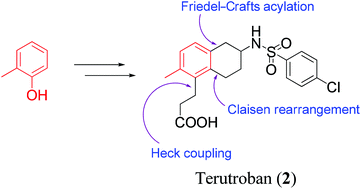
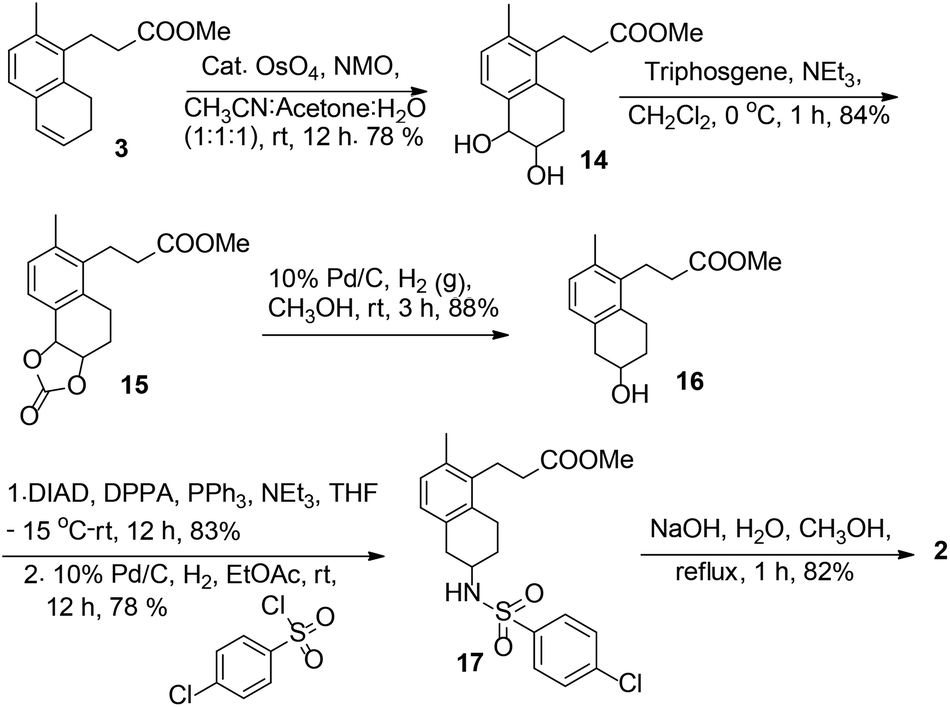
![3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid NMR spectra analysis, Chemical CAS NO. 165538-40-9 NMR spectral analysis, 3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid H-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-09/001/775/1775661_1h.png)
![3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid NMR spectra analysis, Chemical CAS NO. 165538-40-9 NMR spectral analysis, 3-[(6R)-6-[(4-chlorophenyl)sulfonylamino]-2-methyl-5,6,7,8-tetrahydronaphthalen-1-yl]propanoic acid C-NMR spectrum](http://pic11.molbase.net/nmr/nmr_image/2014-11-09/001/775/1775661_13c.png)





)/Images/CSIRlogo.gif)
)/Images/iictlogo.gif)
)/Images/iict70logo.gif)
 DR AV RAMA RAO
DR AV RAMA RAO Professor J. R. Falck
Professor J. R. Falck Professor L. F. Tietze
Professor L. F. Tietze



 .Prof. Dr. Beate Koksch
.Prof. Dr. Beate Koksch fu-berlin.de
fu-berlin.de .
.
 ++49 – 30 – 838 55344
++49 – 30 – 838 55344
 e-mail
e-mail
 homepage
homepage

") An Anopheles female mosquito that transmits malaria(© picture alliance/dpa Fotografia)Over one million people die of malaria each year because they do not have access to effective drugs.Millions, especially in the developing world, cannot afford the combination drug preparation, which consists mainly of artemisinin.
An Anopheles female mosquito that transmits malaria(© picture alliance/dpa Fotografia)Over one million people die of malaria each year because they do not have access to effective drugs.Millions, especially in the developing world, cannot afford the combination drug preparation, which consists mainly of artemisinin.") Dr. Peter H. Seeberger, Director at the Max Planck Institute of Colloids and Interfaces in Potsdam and Professor of Chemistry at the Freie Universität Berlin(© dpa)“Photochemistry is a simple and cost-effective method. However, the pharmaceutical industry has not used it to date because it was so difficult to control and implement on a large scale,” explains Peter Seeberger.
Dr. Peter H. Seeberger, Director at the Max Planck Institute of Colloids and Interfaces in Potsdam and Professor of Chemistry at the Freie Universität Berlin(© dpa)“Photochemistry is a simple and cost-effective method. However, the pharmaceutical industry has not used it to date because it was so difficult to control and implement on a large scale,” explains Peter Seeberger.



 h
h .
.